
The effect of redox properties of ceria-supported vanadium oxides in liquid phase cyclohexene oxidation
Abstract
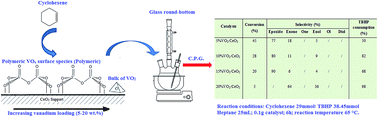
Several VO2/CeO2 based catalysts, with V loadings between 5 and 20 wt%, were prepared by impregnation and subsequently dried, calcined and reduced at 673 K. The structural characterization of these materials was carried out using X-ray diffraction (XRD), N2 adsorption–desorption at 77 K, UV-vis diffuse reflectance spectroscopy (DR UV-vis) and Fourier-transform infrared spectroscopy (FTIR). Their catalytic activities in the epoxidation of cyclohexene, with TBHP as oxidant and heptane as solvent, were also examined. XRD results revealed a good dispersion of VO2 species on ceria. FTIR and DR UV-visible spectroscopy techniques indicated that polymeric vanadyl species are present on all catalysts and a crystalline VO2 phase appears on the 20 wt% vanadium sample. Experimental results revealed that catalyst activity decreases with increased vanadium loading, whereas the epoxide selectivity increased up to 90% when the vanadia loading reached 15 wt%. In the case of 20 wt% VO2/CeO2, the reaction switched towards a 100% enol, enone selectivity. The presence of CeVO4 was shown to influence the conversion and selectivity of the reaction.
 Please wait while we load your content...
Please wait while we load your content...

