
Suppression of coke formation and enhancement of aromatic hydrocarbon production in catalytic fast pyrolysis of cellulose over different zeolites: effects of pore structure and acidity
Abstract
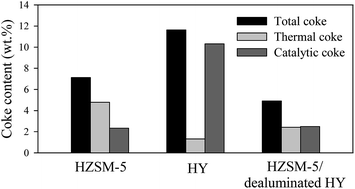
Rapid deactivation of zeolites caused by high formation and deposition of coke is a great challenge in catalytic conversion of biomass materials into value-added chemicals and fuels. The main purpose of this work was to reduce the formation of both types of thermal and catalytic coke over zeolites. It was revealed that there is a significant interaction between zeolite pore structure and the density of acid sites which could be optimized for reduced coke formation. In this study, catalytic pyrolysis of cellulose was conducted using HZSM-5 (Si/Al: 30), HY (Si/Al: 30) and physically mixed catalysts of HZSM-5 (Si/Al: 30) and dealuminated HY (Si/Al: 327). Coke formation over the physically mixed catalysts was remarkably lower than that over HZSM-5 and HY; the coke contents of HZSM-5, HY and physically mixed catalysts of HZSM-5 and dealuminated HY with a ratio of 70 : 30 wt% were 7.01, 11.47 and 4.82 wt%, respectively. The aromatic hydrocarbon yield was also considerably enhanced over the physically mixed catalysts compared to HZSM-5 and HY; the aromatic hydrocarbon yields achieved over HZSM-5, HY and the mixture of HZSM-5 and dealuminated HY (70 : 30 wt%) were 20.31, 8.91 and 27.01 wt%, respectively. This study shows that the interactive effects of zeolite characteristics such as pore structure and acidity could be taken into account for designing more efficient catalysts to achieve lower coke formation and higher production of desired products.
 Please wait while we load your content...
Please wait while we load your content...

