
Titanate and titania nanostructured materials for environmental and energy applications: a review
Yanyan Zhanga,
Zhelong Jiangac,
Jianying Huangb,
Linda Y. Limad,
Wenlong Lia,
Jiyang Denga,
Dangguo Gonga,
Yuxin Tang*a,
Yuekun Lai*b and
Zhong Chen*a
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore. E-mail: tang0212@ntu.edu.sg; ASZChen@ntu.edu.sg
bNational Engineering Laboratory for Modern Silk, College of Textile and Clothing Engineering, Soochow University, Suzhou 215123, China. E-mail: yklai@suda.edu.cn
cDepartment of Materials Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
dDepartment of Materials Science and Engineering and Department of Chemistry, Stanford University, Stanford, California 94305, USA
First published on 11th September 2015
Abstract
Nanosized TiO2-based materials with unique structural and functional properties have already led to breakthroughs in various applications including photocatalysis, adsorption, lithium-ion batteries, etc. In this review, we present the state-of-the-art development of fabrication strategies of titanate/titania nanostructures and their corresponding environmental and energy applications. First, the structural features of titanate and titania and their correlation are explained in great detail. After which, recent research efforts on the development of multi-dimensional titanate materials are summarized. Following that, the applications of titanate/titania nanomaterials in the fields of adsorbents, photocatalysis, lithium-ion batteries, photovoltaics, electrochromic devices, self-cleaning and oil–water separation are reviewed. Finally, the future perspectives for the nanostructured titanate and titania are discussed. Continuous development in this area is essential to endow TiO2-based materials with advanced functionality and improved performance for practical applications.
1. Introduction
Problems concerning energy depletion and environmental pollution have become hot topics across the globe in recent decades.1,2 Our rapacious consumption of carbon-based fossil fuel energy sources has led to carbon dioxide accumulation in the atmosphere, with the outcome of global warming and climate change. Although in danger of depletion, fossil fuels have continued to be the major source of energy driving human civilization.3 The energy and environmental concerns call upon the reformation of our energy structure to rely on green and sustainable sources, such as solar energy. In addition, air and water, the most fundamental matter that life depends on, have become threats to humans due to the discharge of waste chemicals. For instance, 1.4 million km2 of China was covered by a hazardous dense haze in January 2013, with more than 800 million victims.4 Effluent water is often contaminated with organic dyes that are hardly bio-degradable. The Fukushima incident in Japan during March 2011 released radioactive ions to the environment. With increased social impact and general public awareness, new materials or approaches are in urgent demand to solve or mitigate these problems.Titanium dioxide is one of the most heavily investigated oxide materials in addressing energy and environmental crises. Due to its favorable electronic and optoelectrochemical properties, it has been widely applied to solar cells, photocatalysts, lithium ion battery electrodes, smart coatings, etc.5–13 However, improved properties are necessary to meet high demand and complex requirements in the markets. Numerous efforts has been made to develop nanostructured titania materials with engineered compositions, morphologies or heterostructures. The prosperous development of titanium dioxide nanomaterials has also thrived the investigation of a class of TiO2-based structures: layered titanate materials.14
Layered titanate has close structural resemblance to titanium dioxide, both composed of TiO6 octahedra units connected by sharing corners and edges. In fact, the distinction between layered titanate and TiO2 is not very clear, especially in the early literature, and the term titanium dioxide is still occasionally used to name the material that should more formally be called layered titanate. Layered titanate materials have attractive features of their own, including extremely large ion-exchange capacity, fast ion diffusion and intercalation, and high surface charge density. These favorable properties of layered titanate are endowed by their unique crystal structure, where the negatively charged two-dimensional titanium-containing sheets are separated to a large distance by exchangeable cations and molecules in the interlayer. Another attractive feature of titanate is its ability to transform to titanium dioxide. This allows the use of titanate as flexible precursors for TiO2 nano-engineering. The ability of fabricating titanate in a variety of shapes allows the morphology to be inherited by the subsequent TiO2 formation. The open crystal structure of layered titanate permits easy and uniform doping of atoms or molecular assembly, which is hard to achieve if working directly with the more compact TiO2 polymorphs. Several reviews summarized the recent development of titanate nanotubes or the derived titania for its photocatalytic application.15,16 In this review, we discuss the recent development of multi-dimensional titanate materials by the newly emerged technologies and their related applications, and give the readers a full picture in terms of the history and technology evolution trends of titanate materials in this area. We first provided a detailed discussion on the crystal structures of this class of layered titanate materials. The synthesis method for titanate formation is then reviewed, ranging from conventional solid state synthesis, to the recently developed approaches that cover titanate with one-dimensional (1D) linear (tube, wire, or belt), 2D sheet, and hierarchical 3D structures. Lastly, the applications of layered titanate materials dealing with energy and environment problems are summarized. Special focus has been given to their use as adsorbent, photocatalyst, battery electrode, photovoltaics cell, electrochromic device, and in self-cleaning, oil–water separation and biomedical applications.
2. Crystal structure of titania and titanate
Titania (TiO2) can exist in a large number of different crystalline structures, but it is generally described as having three distinct polymorphs: rutile, anatase, and brookite (Fig. 1). In all polymorphs, titanium cations are six-fold coordinated to oxygen anions, forming distorted TiO6 octahedra, and all polymorphs have TiO6 octahedra joined by sharing the octahedral edges (some have corner sharing as well). The different crystal structures of titania differ by the spatial arrangement of the TiO6 octahedra building blocks. Rutile is the thermodynamically stable form of bulk titania, which has tetragonal structure with a space group P42/mnm (136) (Fig. 1a). Although being meta-stable at the bulk form, anatase and brookite can be stable when the crystal is in nano-size, due to their smaller surface energy. Comparing among equal sized particles, Zhang et al.17 employed thermodynamic analysis and suggested that anatase, brookite, and rutile are stable when the size is less than 11 nm, from 11 nm to 35 nm, and greater than 35 nm, respectively. Obviously, the shape and size of nanoparticles will be affected by the experimental conditions (synthesis method, temperature, duration, precursor, etc.). In the work by Zhang et al.,17 they found that when the heating temperature was at 450 °C, only anatase and brookite were present in the mixed phase particles, and their sizes varied from around 10 to 14.5 nm, and from around 12 to 17 nm respectively, when heating time increased from 2 to 24 h. Rutile phase began to form after heating the anatase and brookite mixture at 580 °C, and the rutile particle grew to around 60 nm after 21 h.17 Tay et al.18 hydrothermally synthesized cubic shaped anatase nanoparticles in size ranging from 18 to 42 nm using TiS2 precursor in 0.5 M NaOH at 200 °C for 24 hours. Anatase also possesses a tetragonal structure, with space group I41/amd (141) (Fig. 1b). Brookite, on the other hand, is orthorhombic with space group Pbca (61) (Fig. 1c). Through hydrothermal synthesis, Tay et al.18 obtained pure brookite nanoplates after treating TiS2 in 1.2 M NaOH at 200 °C for 24 hours. The length of the nanoplates was around 100–400 nm, the width and thickness were in the range of 40–110 nm and 10–30 nm, respectively. | ||
| Fig. 1 Crystal structures of TiO2 polymorphs: (a) rutile; (b) anatase; (c) brookite; and (d) TiO2(B). Purple spheres represent Ti atom, and the blue octahedra represent TiO6 blocks. Oxygen atoms at the corner of the octahedra are omitted for clarity. | ||
In addition to rutile, anatase, and bookite, there is one exotic phase of pure titania, TiO2(B). This phase is often formed via solution synthesis. Although having the same chemical formula TiO2, TiO2(B) possesses a very open crystal structure (Fig. 1d) and shares more similarity with titanate (to be elaborated later) than other TiO2 phases. Its space group C2/m (14) is the same as monoclinic titanate (Fig. 2a), and can be imagined of as titanate sheet having structural entity composed of only two TiO6 octahedra, with adjacent sheets joined together by edge-sharing. Other than precipitating directly from solution, TiO2(B) was identified as an intermediate product in the calcination of titanate, following the transformation sequence of protonated titanate → TiO2(B) → anatase TiO2.19 Anatase and rutile are the most studied polymorphs of titania for solar driven applications such as photocatalyis and photovoltaics. Both of them have indirect band gap, which is 3.0 eV for rutile and 3.2 eV for anatase, corresponding to ultra-violet (UV) light absorption. It is generally accepted that anatase is the most active phase for photocatalysis applications, due to its better electronic and surface chemistry properties. Their properties can be further improved via doping or forming heterojunctions with other phases with favorable electronic coupling.
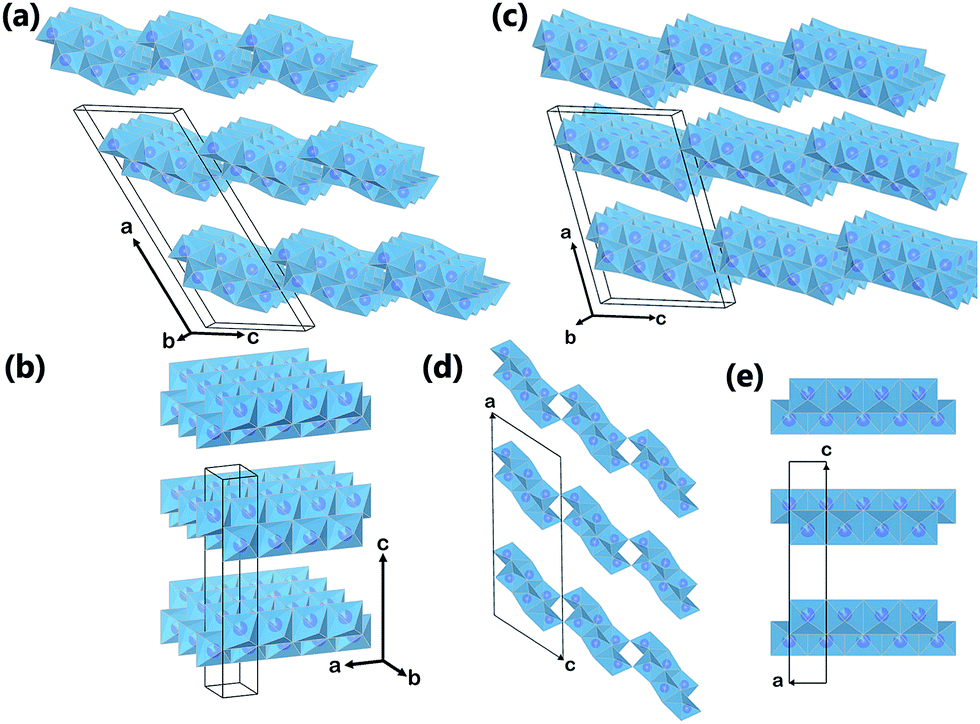 | ||
| Fig. 2 Crystal structures of (a) monoclinic titanate H2Ti3O7; (b) orthorhombic titanate HxTi2−x/4□x/4O4; (c) monoclinic titanate H2Ti4O9. The (010) plane projection of (d) monoclinic titanate H2Ti3O7, and (e) orthorhombic titanate. Purple spheres represent Ti atom, and the blue octahedra represent TiO6 blocks. Oxygen atoms at the corner of the octahedra, other cations and molecules in the interlayers are omitted for clarity. | ||
Other forms of TiO2 and TiO2-based phases exist. In their early discovery, these TiO2-based materials have been mistakenly assigned as anatase TiO2.20,21 However, later investigations confirmed that they were actually a class of layered titanate material, with a general formula MxTiyOx/2+2y·zH2O (M = H, Li, Na, K, etc.).22,23 These titanate structures have 2D layers formed by connected TiO6 octahedral blocks, with cations and neutral molecules inserted into the inter-layer space. Based on crystal symmetry, the titanate materials can be further divided into two subclasses: monoclinic and orthorhombic (Fig. 2a and b). All monoclinic forms of titanate are found to belong to space group C2/m (12). The 2D sheets in monoclinic titanate are zigzag in nature. Each of the zigzag layers is built upon the structural entity composed of several collinear edge-sharing TiO6 octahedra lying in the (010) planes. Every single structural entity is joined by corner to the other structural entities related to it by c-translation, and is further joined by edge to other structural entities related to it by 21 screw operation (at a = ±1/4, c = 1/2), forming a continuous 2D sheet. The various stoichiometry of monoclinic titanate is related to the number of TiO6 octahedra composing the structural entity. For example, in H2Ti3O7, the structural entity is composed of 3 octahedra blocks (Fig. 2a), whereas in H2Ti4O9, the structure entity has 4 octahedra blocks (Fig. 2c). The layering nature monoclinic titanate can also be influenced by the size and amount of the other cations relative to titanium. When there is not enough cations to support the large interlayer spacing, the separate interlayers will join together by sharing TiO6 octahedra corners. This can be taken advantage of for the treatment of radioactive ions, as will be discussed later. The other form of layered titanate has an orthorhombic crystal structure with space group Immm (71), which is frequently termed lepidocrocite titanate (for their structural analogy to γ-FeOOH) (Fig. 2b).24 Although being in a different crystal system, the TiO6 octahedra arrangement in orthorhombic titanate is closely related to monoclinic titanate. Indeed, the orthorhombic titanate can be visualized as the monoclinic titanate with the structural entity composed of infinite number of TiO6 octahedra. In effect, orthorhombic titanate layer is straight and continuous in the a-direction. Because of the absence of corner-sharing, charge balance have to be maintained from partial occupancy of Ti sites, leading to chemical formula HxTi2−x/4□x/4O4·zH2O (where □ denotes vacancy, and x is close to 0.7).25 Distinction between the monoclinic and orthorhombic titanate is usually difficult, because their crystallinity is usually low, and their structural similarity resulted in close characteristic peak positions in XRD. Projections of monoclinic titanate H2Ti3O7 and orthorhombic titanate onto their (010) place is illustrated in Fig. 2d and e.
Thanks to the open crystal structure and large interlayer spacing in titanates, they can accommodate many varieties of cations and neutral molecules in the interlayer spaces, endowing them unique physiochemical properties. Large dipole moment exists in titanate materials, where the negatively charged titanate layer is balanced by positively charged interlayer cations. Layered titanate has very good cation exchange capability. Because of the negative charge on the titanate sheet, it can adsorb positively charged molecular species effectively. These effects have been applied in waste water remediation for the treatment of radioactive ions and organic dyes, which is the subject of Section 4.1. Although titanate in its native form is not very effective for solar-driven applications, it can be transformed to titania via calcination with minimal morphological distortions. Therefore, titanate can serve as precursor and scaffold for more active materials. The ability to modify the content within the interlayer of titanate provides a means for doping or forming hetero-structures of titania. Since the dopant is mixed at an atomic level through the interlayer, the formed TiO2 by transformation of modified titanate is expected to have uniform doping, in contrast with the shallow surface doping by diffusion in bulky materials. The potential of titanate in the area of photocatalysis will be discussed in Section 4.2. The open framework of titanate also provides fast Li intercalation pathways, making it a candidate cathode material for Li-ion battery, which will be addressed in Section 4.3. Section 4.4 highlights applications of TiO2 for photovoltaic and electrochromic devices. Other functionalities of titanate stem from its surface hydrophilicity and the ability to turn to hydrophobicity through chemical modification, making them useful in biomedical application, self-cleaning and oil–water separation applications, are to be discussed in Section 4.5.
3. Development of multi-dimensional titanate materials
In this section, the technological development for the formation of multi-dimensional titanate materials will be described. The formation conditions of titanate include the followings: (1) having titanium precursor sources (TiO2, Ti, etc.); (2) being in alkaline reaction environment; (3) negative free energy change (i.e. thermodynamically spontaneous) for the reaction.
 | (1) |
Based on this guidance, the experimental designs toward multi-dimensional (bulk, 0D, 1D, 2D, and 3D) titanate materials by solid state method, hydrothermal reaction, electrochemical spallation, high-temperature oxidation, and other approaches have been developed, and the history of key development of titanate materials are summarized in Fig. 3. Aiming at achieving better functional properties as well as improved performance, there has been a strong incentive for the size and morphology control of titanate materials.26
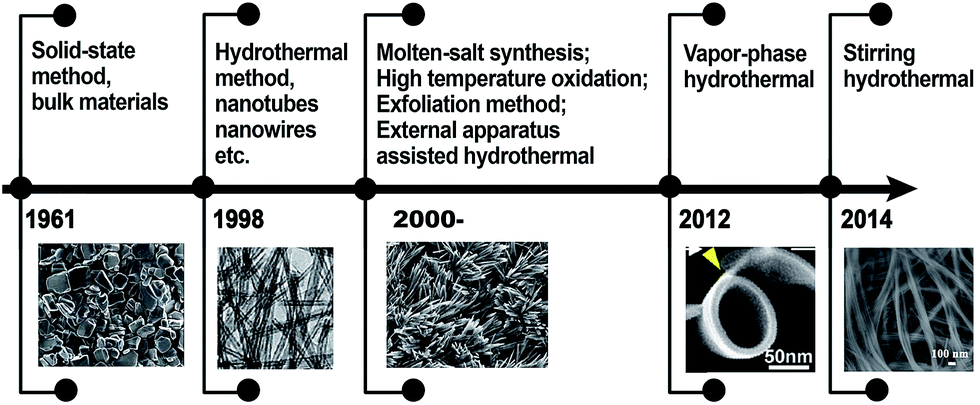 | ||
| Fig. 3 The history of technological development in the synthesis of titanate materials. | ||
3.1. Fabrication of bulk titanate materials by solid-state reaction
Solid state reaction refers to a chemical process without the usage of solvent. The reaction usually takes place at a high temperature for several days to ensure the complete reaction. The typical titanate formula A2TinO2n+1 consists of an alkali metal oxide (A2O) and titanium dioxide (nTiO2). Therefore, different kinds of bulk titanate materials can be obtained by mixing TiO2 with the metal carbonate (A2CO3, A = Cs, K or Na, etc.) with certain molar ratio. Due to evaporative loss of the alkaline component at the calcination temperature, the final product will not be a single phase of A2TinO2n+1 if starting from a stoichiometric mixture. Therefore, the experimental molar ratio of the alkali oxide and the titanium dioxide should be higher than calculated stoichiometry.Researchers have synthesized different kinds of alkali metal titanates A2TinO2n+1. For example, the Na2Ti3O7 and K2Ti4O9 were synthesized by heating mixtures of Na2CO3 and TiO2 mixture (≤1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 molar ratio), K2CO3 and TiO2 mixture (1:3.5 molar ratio) at 800 °C for 20 h.27 With the same approach, Sasaki et al.28 used Cs2CO3 and TiO2 (1:4.5 molar ratio) as starting reagents, synthesized the bulk Cs2Ti5O11 material. The protonation process (hydrogen-exchange) of Cs2Ti5O11, Na2Ti3O7 and K2Ti4O9 were performed in the acid solution. The acid solution was refreshed frequently to completely remove alkali ion from the compounds, leading to the final products of protonated titanate H2Ti5O11, H2Ti3O7 and H2Ti4O9 respectively. Moreover, the starting material of the alkali carbonate (A2CO3) can also be replaced by ANO3, AOH or A2O (A = Cs, K or Na, etc.) and so on. For example, Tournoux and co-workers29 heated KNO3 and TiO2 at 1000 °C for 48 h and the product was K2Ti4O9. The resultant powder was then hydrolyzed with HNO3 at room temperature to yield a solid product H2Ti4O9·H2O. Wang et al.30 also adopted a similar method to synthesize K2Ti4O9 using K2O and TiO2 as the starting materials.
:3 molar ratio), K2CO3 and TiO2 mixture (1:3.5 molar ratio) at 800 °C for 20 h.27 With the same approach, Sasaki et al.28 used Cs2CO3 and TiO2 (1:4.5 molar ratio) as starting reagents, synthesized the bulk Cs2Ti5O11 material. The protonation process (hydrogen-exchange) of Cs2Ti5O11, Na2Ti3O7 and K2Ti4O9 were performed in the acid solution. The acid solution was refreshed frequently to completely remove alkali ion from the compounds, leading to the final products of protonated titanate H2Ti5O11, H2Ti3O7 and H2Ti4O9 respectively. Moreover, the starting material of the alkali carbonate (A2CO3) can also be replaced by ANO3, AOH or A2O (A = Cs, K or Na, etc.) and so on. For example, Tournoux and co-workers29 heated KNO3 and TiO2 at 1000 °C for 48 h and the product was K2Ti4O9. The resultant powder was then hydrolyzed with HNO3 at room temperature to yield a solid product H2Ti4O9·H2O. Wang et al.30 also adopted a similar method to synthesize K2Ti4O9 using K2O and TiO2 as the starting materials.
Although different kinds of bulk titanate materials have been prepared through solid state reaction, the resultant particle sizes of these prepared materials are large, and the specific surface areas are small, which limits the materials' practical application in photocatalysis and a number of other areas. In order to obtain high photocatalytic activity to or improve ion-exchange/intercalation performance, it is necessary to synthesize nano-sized materials possessing large specific surface areas and with special morphological features. On the other hand, the bulk titanate materials can be precursor for the formation of nanosheets by exfoliation method, which is discussed in the following sections.
3.2. Controlled growth of 0D titanate/titania nanoparticles
0D nanoparticle refers the ones with spherical or nearly equal-axial dimensions within the nanosize range (<100 nm). 0D titanate nanoparticles are rarely reported in the past decades, probably due to the difficulty in experimental control. Our group31 reported a sol–gel method combined with solvothermal approach to fabricate titanate/titania hybrid nanoparticles. These particles have a high surface area (180 m2 g−1) and the transmission electron microscopy (TEM) image (Fig. 4a) shows cubic particles of averaging 20 nm in size, with an internal structure of tessellation suggestive of intra-granular pores with a size around 3.0–3.5 nm. HRTEM image is shown in Fig. 4b of a single particle consisting of both anatase (cyan color) and titanate (red color) nanocrystallites that share intimate interface. Each phase has an average crystal size around 5 nm. A fast Fourier transformation (FFT) study of the entire area shows two sets of diffraction patterns (see inset of Fig. 4b). The detailed formation mechanism is not fully understood. We suspect that the sol–gel approach can provide homogenous titanium source precursors, and the following solvothermal approach promotes crystallization process of titanate nanoparticles in alkaline condition (in triethylamine) via a bottom-up strategy. | ||
| Fig. 4 (a) TEM and (b) HRTEM image of the as-synthesized photocatalyst with two phases distinguishable by color. The inset shows the diffraction pattern obtained from the FFT study of the monoclinic titanate (weak spots) and tetragonal anatase phases (strong spots).31,32 | ||
Recently, titanate-derived TiO2(B) nanoparticles33 were prepared using a hydrothermal method followed by thermal treatment. Firstly, a titanium glycolate complex was formed by dissolving Ti metal in a mixture of H2O2 and NH3 in water. Then the mixture is subjected to hydrothermal treatment, forming titanate nanoparticles. Then the as-prepared product is calcined in dry air to form TiO2(B). The TEM data demonstrates that the material is composed of nanoparticles of ca. 2.5–4.3 nm size (based on analysis of 100 nanoparticles), with a relatively narrow size distribution (Fig. 5). These nanoparticles form agglomerates of 0.3–3.0 μm in size. BET surface area determined from N2 adsorption is 251 m2 g−1 (pore volume 0.12 cm3 g−1).
 | ||
| Fig. 5 HRTEM image of TiO2(B) nanoparticles. White boxes delineate primary (nano) particles within the agglomerates.33 | ||
3.3. Rational synthesis of 1D titanate nanostructures
 | ||
| Fig. 6 Titanate nanotubes: (a and b) HRTEM image; (c) enlarged HRTEM image of a part in (a); (d) structure model of one unit cell of H2Ti3O7 on the [010] projection; (e) schematic drawing of the structure of nanotubes; (f) three-dimensional drawing of a nanotubes.34 | ||
However, whether the crystal structure of titanate nanotubes belongs to monoclinic or orthorhombic system is still under the debate. This is caused by a few reasons: (1) its crystallite size is very small, which leads to the broadening of peaks in the XRD patterns; (2) the proposed crystal structures share close similarities; (3) the precise positions of hydrogen atoms inside the crystals is difficult to be located.15 The trititanate acid (H2Ti3O7), lepidocrocite type titanate (HxTi2−x/4□x/4O4, □ represents vacancy), dititanate acid (H2Ti2O5·H2O), and tetratitanate acid (H2Ti4O9·H2O) had been proposed as shown in the Table 1. Peng and co-workers34 proposed that the crystal structure of titanate nanotubes corresponded to the layered H2Ti3O7 with a monoclinic crystal structure, which is obtained by rolling several (100) planes around axis [010] or [001]. However, Ma et al.24 suggested that the titanate nanotube possessed a lepidocrocite-type (H0.7Ti1.827□0.175O4·H2O, where □ indicates a vacancy) structures, which is supported form the selected area electron diffraction study. According to the Raman study, Gao et al.35 believed that the crystal structure of titanate nanotube could be better described by protonated titanate H0.7Ti1.827□0.175O4·H2O, as it has been demonstrated that the different types of polymerization of TiO6 octahedra in these compounds result in the different vibrational features. Lepidocrocite titanate contains “flat” host layers in which TiO6 octahedra are combined with each other via edge-sharing, while the crystal structure of H2Ti3O7 consists of corrugated “ribbons” of edge-sharing TiO6 octahedra, where the octahedra join at the corners to form a “stepped” layered structure, as shown in Fig. 2.
| H2Ti3O7a | H2Ti4O9·H2Ob | H2Ti2O5·H2Oc | H0.7Ti1.825□0.175O4·H2Od | ||||
|---|---|---|---|---|---|---|---|
| d | h k l | d | h k l | d | h k l | d | h k l |
| a JCPDS 47-0561 monoclinic; lattice constants a = 16.023, b = 3.749, c = 9.191, and β = 101.45°.b JCPDS 36-0655 monoclinic; a = 18.77, b = 3.75, c = 11.62, and β = 104.6°.c JCPDS 47-0124 orthorhombic; a = 18.03, b = 3.783, and c = 2.998.d Calculated from JCPDS 83-0702 orthorhombic; a = 3.783, b = 18.545, and c = 2.982. | |||||||
| 7.87 | 2 0 0 | 9.05 | 2 0 0 | 9.04 | 2 0 0 | 9.272 | 0 2 0 |
| 3.65 | 1 1 0 | 3.672 | 1 1 0 | 3.696 | 1 1 0 | 3.707 | 1 1 0 |
| 3.05 | 3 1 0 | 3.185 | 3 1 0 | 3.204 | 3 1 0 | 3.226 | 1 3 0 |
| 2.67 | 3 1 −2 | 2.668 | 2 1 −3 | 2.684 | 3 0 1 | 2.686 | 0 3 1 |
| 2.37 | 1 1 −3 | 2.298 | 2 1 −4 | 2.306 | 5 0 1 | 2.324 | 0 5 1 |
| 1.88 | 0 2 0 | 1.876 | 0 2 0 | 1.893 | 0 2 0 | 1.891 | 2 0 0 |
| 1.478 | 2 0 2 | 1.491 | 0 0 2 | ||||
Since its introduction,20 the alkaline hydrothermal production of titanate nanotube has been engineered in a direction of attaining the ease of operation, diminishing process cost, and facilitating scale-up. Further development of this alkaline hydrothermal strategy has revealed that all TiO2 polymorphs (including anatase, rutile, amorphous TiO2, or even Ti metal) can be employed as the precursor in the formation of nanotubular or nanofibrous TiO2 (Fig. 7).14 Some classic preparation processes for the fabrication of titanate nanotubes are shown in Fig. 7, which includes the original method (route 1 in Fig. 7) of dispersing TiO2 raw material in solutions concentrating 10 M NaOH at 110–150 °C temperature range for a duration of 24 h. Recently, it has been reported that using alternative solvents, the synthesis of titanate nanotube can be achieved at lower temperatures. For instance, using a mixture of NaOH and KOH36 (route 2 in Fig. 7), the conversion can be brought into near completion at a temperature as low as 100 °C with the aid of refluxing atmospheric pressure. Additional improvement in this reflux strategy includes further optimization of the aqueous solution composition and the addition of additives in order to synthesize titanate nanotubes at even lower temperatures for shorter durations.
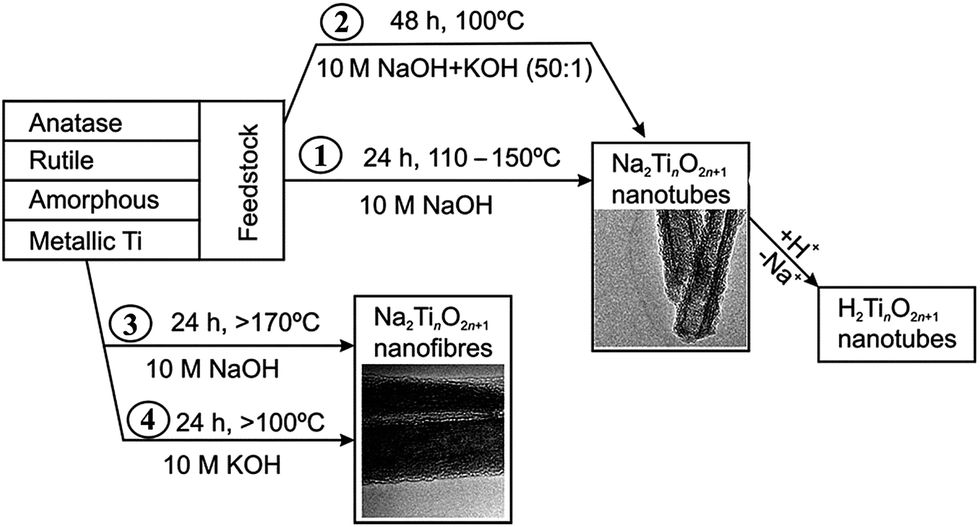 | ||
| Fig. 7 Respective routes to the manufacture of titanate nanotubes and nanofibers.37 | ||
The formation of titanate nanofibers, wires or belts usually employs hydrothermal treatment of TiO2 in 10 M NaOH solution at higher temperatures than the case of nanotube (>170 °C) (route 3 in Fig. 7).38 For example, as shown in Fig. 8, Zhu's group39 reported that the titanate nanowires were formed at 180 °C for 48 h by using the rutile titania as the starting materials. The formation of nanofiberes can also be achieved if 10 M NaOH is substituted by 10 M KOH solution (route 4 in Fig. 7).40 The decrease of the reaction temperature tend to form nanotubes/nanofibers mixture.41 Our group42 has synthesized titanate nanobelts via a hydrothermal method using titanium disulfide (TiS2) as the precursor. Impressively, sodium titanate nanobelts could be formed under a relatively low alkaline concentration (1 M NaOH) and short duration (6 h).
 | ||
| Fig. 8 (a) Low- and high-magnification TEM images of the nanowires synthesized by treating rutile with 10 M NaOH for 48 h at 180 °C.39 | ||
In brief, hydrothermal conditions, which include the type and concentration of the solution, temperature, and time, play a significant role for the controlled synthesis of titanate nanostructures. For example, Morgan's group43 systematically investigated caustic concentration and temperature on nanostructure formation from Degussa P25 through alkaline hydrothermal treatment. The morphological phase diagram is provided in Fig. 9, which is consistent with the above results and it is a useful guideline for nanostructure morphology control for the titanate materials.
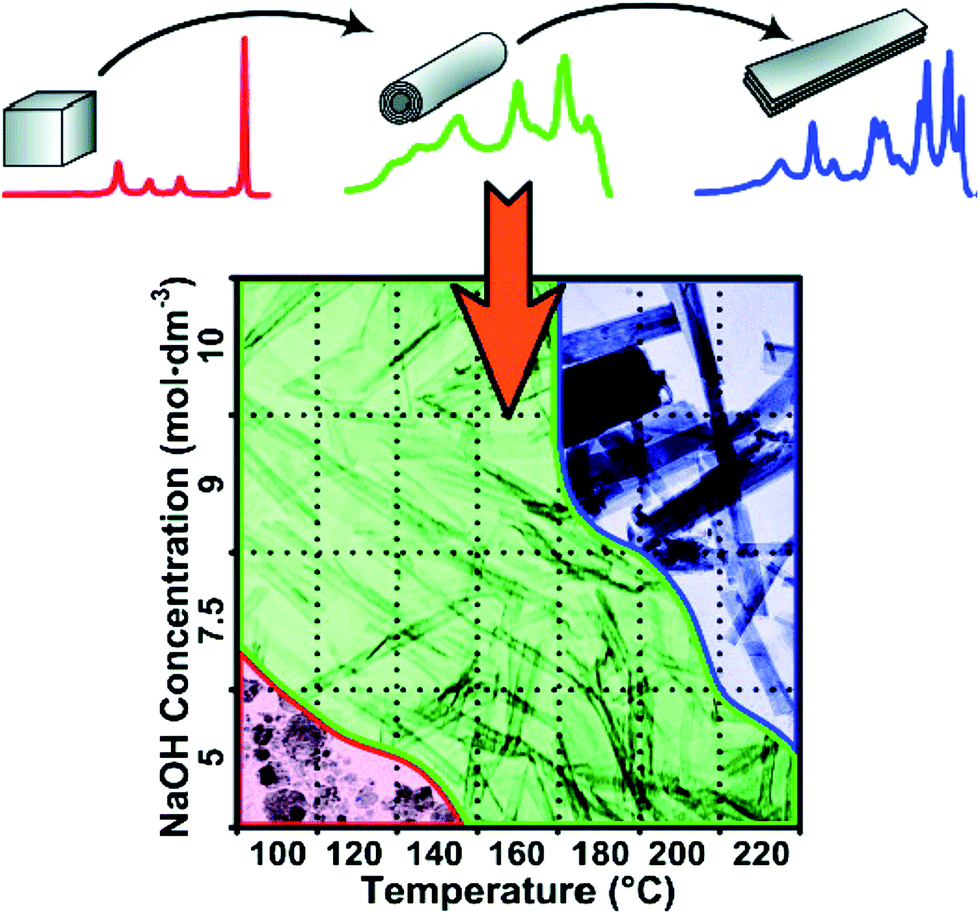 | ||
| Fig. 9 Morphological phase diagram of Degussa P25 evolution indicating regions of nanostructure formation after 20 h of hydrothermal treatment.43 | ||
It is evident that TiO2 with different phase and morphology can be transformed to titanate nanotubes under specific hydrothermal conditions, but its formation mechanism and the detailed transformation for the nanotubular titanates is still unclear. Under hydrothermal condition, the intermediate single-layer or multilayered titanate nanosheets are observed and they play an important role during the formation of nanotubular morphology. The nanotubes can be formed by the scrolling and folding action of the sheets, and the driving force for this curving is proposed by several groups. Zhang's group44 proposed that an asymmetrical chemical environment (Fig. 10a) caused by the H+ or Na+ ion concentration imbalance on the different locations of the titanate nanosheets resulted in the bending of the nanosheets, generating excess surface energies. Bavykin et al.38 proposed the bending of multilayered nanosheets is due to the mechanical tensions (Fig. 10b) resulted from the nanosheets dissolution/crystallization process. The spontaneous crystallization and rapid growth of the layers may result in various layer widths, and it is likely that the tendency of releasing the excess surface energy, which has its root in the layer width imbalance, drives the movement of layers within the multiwall nanosheet. The consequence of this movement is the bending of multilayered nanosheets.14 It is noted that the nanotube axis is along the [010] direction and thus the nanosheets bend around the b-axis due to the imbalance in the nanosheet along the c-axis. When the hydrothermal temperature is increased to 170 °C, the titanate nanofibers or nanowires are formed and axis of the long wire is the [001] direction. At this high temperature, it is proposed that no nanosheets bending should occur and the resulting nanowires grow along the c direction ([001]), which is due to the maximization of dissolution/crystallization rate along this axis.
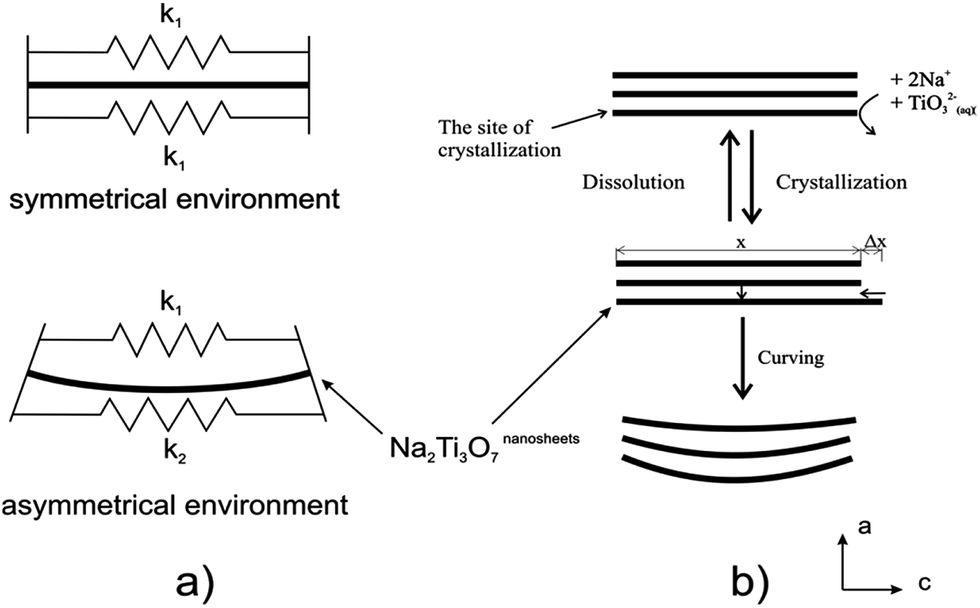 | ||
| Fig. 10 The driving forces for bending titanate nanosheets under the alkaline hydrothermal conditions. (a) Asymmetrical chemical environmental resulting in different surface tensions. (b) Imbalance in layer widths resulting in shifting of the layer and bending the nanosheets.14 | ||
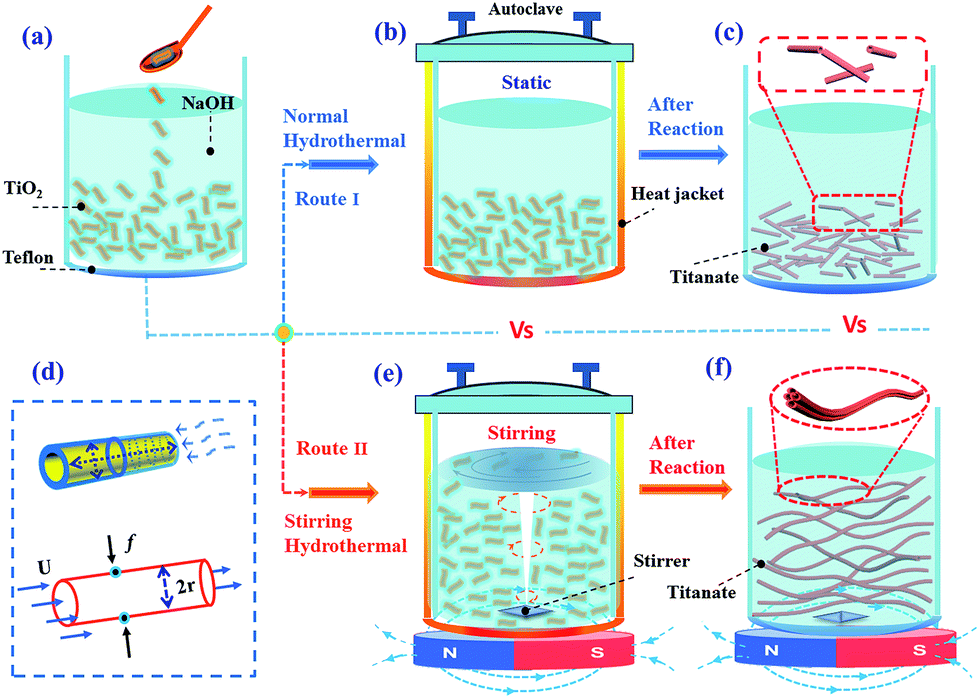
3.3.2.1 External-apparatus assisted hydrothermal method. Although the discussed hydrothermal techniques have demonstrated the potential for the formation of titanate nanostructures, there are still some limitations: (1) slow reaction kinetics, which results in long reaction time; (2) short length of the nanotubes; (3) non-uniformity at large-scale. Various approaches, such as ultrasonication-assisted, microwave-assisted, and rotation-assisted hydrothermal methods have been explored to solve these problems. For example, the sonication and irradiation treatment can influence the morphology of titanate/titania nanostructured materials. The length of titanate nanotube becomes longer with the application of sonication pretreatment.45 Zhu et al.46 found that titanate whiskers and nanotubes were obtained at a sonication power of 580 W and 280 W, respectively. The titanate whiskers are obtained as a slender sheet with a length of about 1 μm and a width of 60 nm, and the nanotubes with a diameter of about 5 nm, and a length of 200–300 nm are obtained. By this sonication–hydrothermal combination approach, Ma et al.47 obtained nanosheet-, nanofiber- and nanotube-like morphologies at a sonication power of 100, 280 and 380 W, respectively. In particular, titanate nanotubes with outer diameters of 5–10 nm and lengths up to 600 nm were obtained. Their study46,47 revealed that the sonication with sufficient power might play an important role that promotes intercalating Na+ into titania lattices and breaking the Ti–O–Ti bonds. This induces easy peeling of large nanosheets, which consequently rolled into longer titanate nanotubes or other morphologies. Moreover, the level of applied microwave irradiation during the fabrication process is responsible for both the intercalation intensity of Na atoms into titanate nanotubes and the type of crystallization phase within nanotubes.48 Interestingly, Dufour et al.49 found that microwave treatment significantly reduces the heating time and generally produces smaller TiO2 nanoparticles, and it also affect the phase composition probably due to the modification of the kinetics of formation of the different phases.
Development of long nanotubes with relatively high surface area would be of great interest to tailor properties for applications.50,51 Recently, Horvath et al. reported conversion of nanotube to nanowire with revolving autoclave (60 rpm) at temperature of 130 °C, which is 20 °C lower than that used in previous literature.52 They suggested that flow pattern created directional mass transfer and facilitated parallel alignment of nanotubes to be further transformed in to nanowires. Inspired by this setup, Torrente et al. synthesized titanate nanotubes with length scale one order of magnitude higher at a lower revolving speed of 20 rpm,53 but agglomeration of nanotubes into secondary linear structure became pronounced. In addition, there are intrinsic limitations associated with such approach including high energy consumption, sophisticated apparatus setup and limited rotation speed achievable (<100 rpm).
3.3.2.2 Stirring hydrothermal method. Although the external-apparatus assisted hydrothermal method has been developed to control the titanate nanotubular structures, the length of the nanotubes was still limited to one-micrometer. To overcome this limitation, our group reported10 a protocol to rationally grow elongated titanate nanotubes with length up to tens of micrometers by a stirring hydrothermal method (Fig. 11). Our detailed study confirmed that the mechanical force-driven stirring process synchronously improved the diffusion and surface reaction rate of titanate nanocrystal growth in solution phase, which is the reason for lengthening of the titanate nanotubes via an oriented attachment mechanism. Besides, such setup also possesses advantageous features including less energy consumption, easier scaling up, and more flexible stirring speed control. Thus it represents a more viable and efficient approach.
 | ||
| Fig. 11 Schematic illustration of the formation process of short and elongated nanotubular structures under normal (a–c) and stirring hydrothermal processes (e and f) at 130 °C for 24 h respectively. (d) The growth model of nanotube along axial and radial direction (top) and the force analysis (down) of an individual nanotube formed in (e).10 | ||
Since the growth process of nanotubular titanate via dissolution–crystallization step is similar to the Ostwald ripening process, we first considered the diffusion-limited Ostwald ripening (DLOR model, Fig. 12), according to Lifshitz–Slyozov–Wagner theory, to be the sole contributor in the growth mechanism. However, the experimental length (L) data of the titanate nanotube fits well with the DLOR model for the short reaction times only (Fig. 12). A remarkable accuracy (R2 = 0.97) of fits over the entire range of experimental data was given by diffusion-limited and surface reaction limited growth (DLSLOR) model. Thus, the growth of the titanate nanotube deviates significantly from the diffusion-limited Ostwald ripening model and follows a mechanism involving both diffusion-control and surface reaction control under the stirring condition.
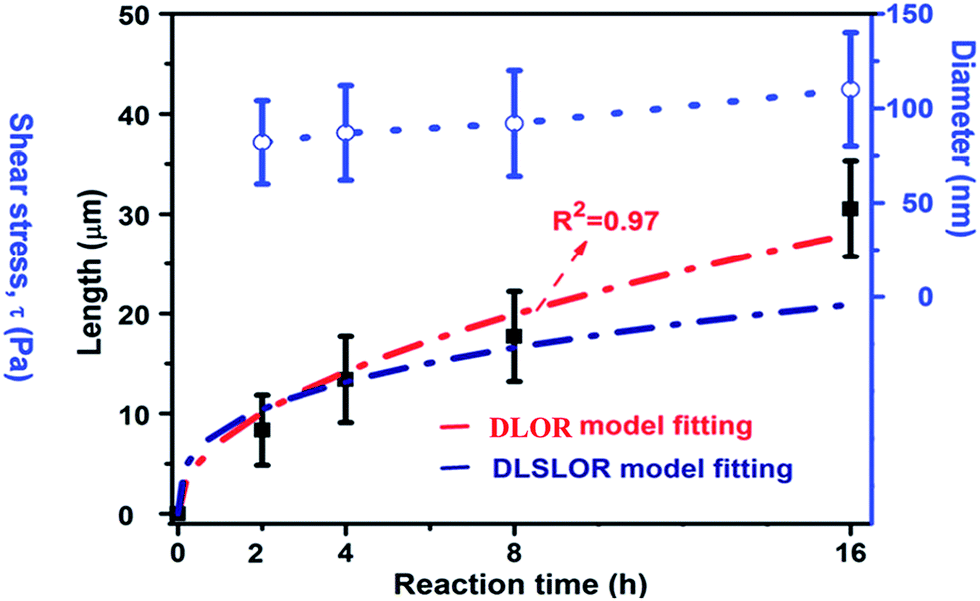 | ||
| Fig. 12 The kinetic study (the nanotubular length, diameter) of the nanotubular sample obtained under 500 rpm. The red curve and navy blue curve fit data L using the mixed diffusion- and surface reaction limited model (DLSLOR model) and diffusion-limited Ostwald ripening (DLOR) control growth model respectively.10 | ||
Based on zero-shear viscosity theory, it is known that in a suspension, solution viscosity (η0) is the sum of the solvent viscosity (ηs) and a contribution from the suspended material:
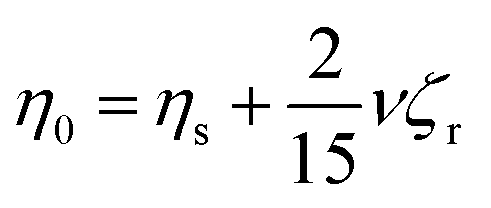 | (2) |
 | ||
| Fig. 13 Fabrication and characterization of titanate nanotubes with different aspect ratios. (a) Digital photos of resulted titanate solution obtained by hydrothermal method at 500 (left) and 0 rpm (right) after sedimentation; (b) FESEM images of titanate nanotubes obtained at 500 rpm; (c) TEM image of (b), the arrow indicating the formation of nanotubular structure; (d) XRD pattern of the products synthesized at different stirring speeds; and (e, f, g, h, i) FESEM images of the titanate nanotubes obtained by hydrothermal reaction at 130 °C for 24 h in at a stirring rate of 0, 300, 400, 500 and 1000 rpm respectively.54 | ||
3.3.2.3 Vapor-phase hydrothermal approach. High-temperature NaOH flux and water vapor were found to play important roles in the growth of uniformly sized titanate nanorods. Recently, vertically aligned titanate nanotubes (Fig. 14) were directly grown on a titanium foil substrate using a vapor-phase hydrothermal approach.44 The resultant nanotubes displayed outer diameters in the range of 50 to 80 nm and average wall thickness of 10 nm, which corresponds to more than 10 titanate layers. It was observed that a distinctive nanosheet rollup mechanism dominated the nanotubes formation process, which differs remarkably from the mechanism frequently encountered in conventional liquid-phase hydrothermal reactions.
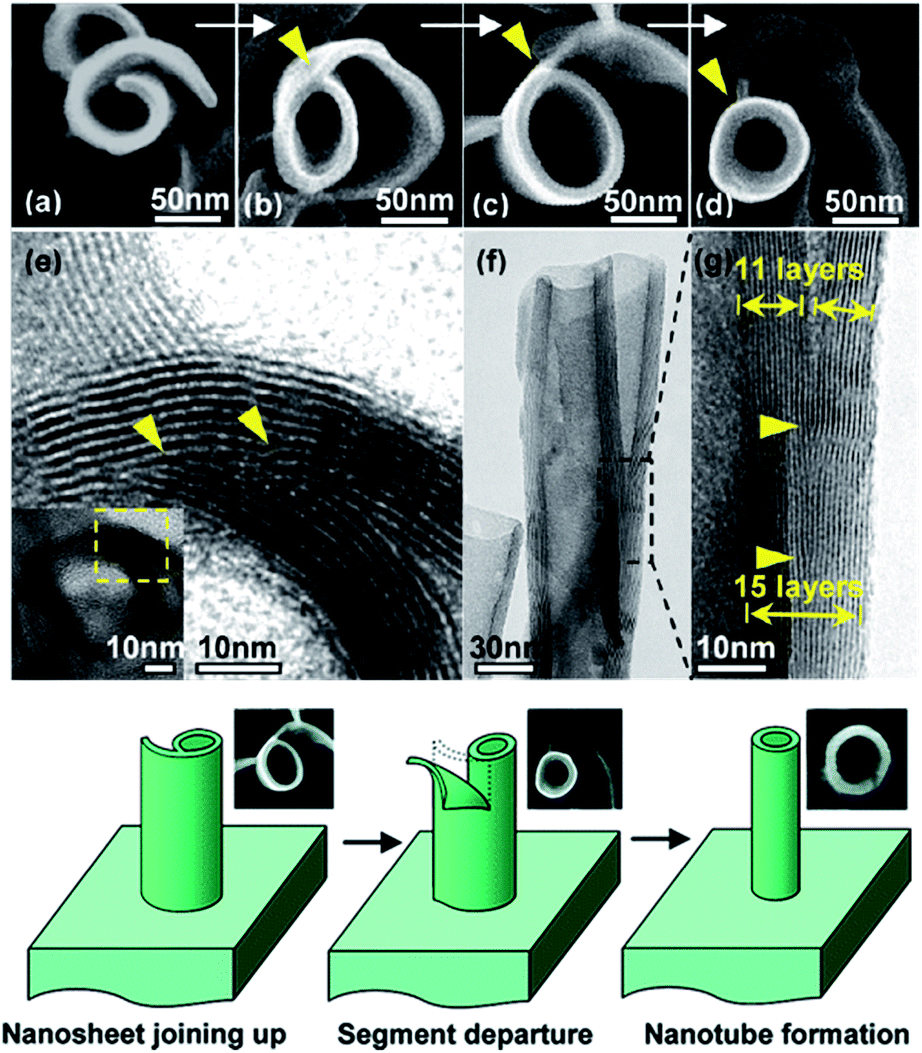 | ||
| Fig. 14 SEM and TEM images of the morphological evolution from a nanosheet into a nanotube; the proposed nanosheet roll-up mechanism is by vapor-phase hydrothermal approach.44 | ||
Beyond the modified hydrothermal method, various approaches have been applied to prepare 1D titanate nanostructures. For examples, the titanate nanowires can also be synthesized from reactions of TiO2 nanoparticles with molten salt in the presence of a nonionic surfactant by a molten-salt synthesis method.55 By this approach, large quantity of nanowires with uniform diameter around 100 nm was obtained with their length ranged from several to a few tens of microns. Moreover, a high-temperature oxidation method to obtain titanate nanorods was reported by Liu et al.56 In this method, NaOH coated Ti metal chips was annealed at 800 °C in the presence of oxygen and water vapor, producing novel tabular titanate nanorods.
3.3.2.4 Sol–gel approach. As a representative of wet chemistry method to fabricate 1D TiO2 nanotubes or nanofibers, sol–gel inherits the merit of low preparation temperature and ease of morphology control. In fact, sol–gel remains the most commonly used method in fabrication of TiO2 (including doped TiO2) for the application of photocatalyst.57 A generic routine to prepare nanocrystalline TiO2 sol–gel formulations is to mix titanium tetraisopropoxide in acetic acid. The mixed acidic solution is further subjected to heating with vigorous stirring to undergo hydrolysis and condensation reaction with the aid of acetic acid to obtain the nanostructured TiO2. The employment of sol–gel titania in corporation with electrospinning was not exploited until 2003, when Xia's group introduced electrospinning to fabricate titania nanofibers.58 Electrospinning is a typical organic fiber fabrication process where polymer solution is injected from a small nozzle under the influence of an electric field. A jet is formed due to the continuous accumulation of electrostatic charges on the surface of the solution droplet. The jet is then subjected to stretching to form ultrathin fiber. By mixing titanium tetraisopropoxide with poly(vinyl pyrrolidone) (PVP) in alcohol solution, Xia's group prepared TiO2/PVP composite nanofibers with a uniform diameter of 78 ± 9 nm. The composite nanofibers were further burned in air at 500 °C to remove PVP residual, leaving titania scaffold nanofibers with a reduced diameter of 53 ± 8 nm. Further modification on the electrospinning has enabled tailoring the nanofibers with core-sheath, hollow or porous structures.59 This design relies on the incorporation of mineral oils loaded into the core capillary while the sol–gel precursor is loaded into the sheath capillary,60 as shown in Fig. 15a. By extracting the mineral oils in the core and removal of PVP in the sheath, robust titania nanotubes were obtained, as shown in Fig. 15b–d. Despite that the combination of sol–gel and electrospinning exhibits straightforward and versatile strategies for synthesizing 1D titania fibers, the fibers are usually randomly aligned. By incorporating recently reported nonelectrospinning spinneret-based tunable engineered parameters (STEP) technique with sol–gel precursor, Wang et al.61 managed to fabricate aligned 1D titania structures including fibers, tubes, nanowires in microtubes. A solution of polystyrene (PS) and titanium tetraisopropoxide was utilized as the precursor. A rotating substrate was employed to pull out the solution filament from the extruded pendent solution droplet and stretch it into micro/nanoscale fibers. After hydrolysis and calcination procedure, highly porous titania hollow tubes with diameters of approximately 250 nm were obtained. Apart from titania, sol–gel process has also seen significant application in fabrication of titanate thin films, such as sol–gel derived lead–zirconate–titanate (PZT) ferroelectronic thin film. Interestingly, study has revealed that the PZT thin films on sapphire substrates was able to complete the pyrochlore to perovskite transformation at 650 °C. As a comparison, free-standing PZT films showed a much higher transformation temperature. The discrepancy in the behavior of free-standing sol–gel derived titanate films versus titanate films on substrate can be related to the size effect of the thin film.62
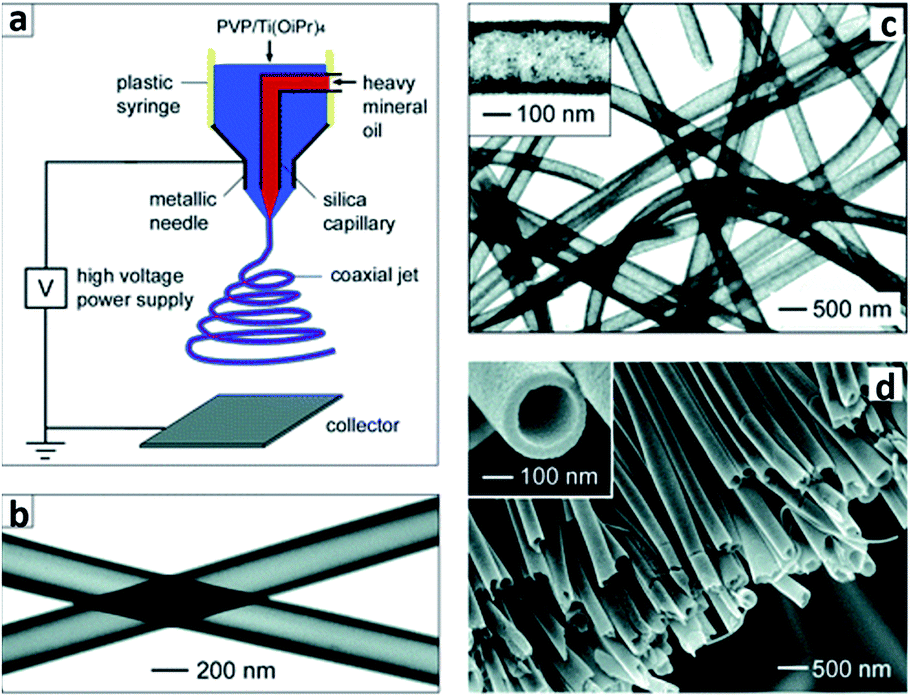 | ||
| Fig. 15 (a) Schematic of the setup for electrospinning nanofibers with a core/sheath structure. (b) TEM image of two as-spun hollow fibers after the oily cores had been extracted with octane. The walls of these tubes were made of a composite containing amorphous anatase TiO2 and PVP. (c) TEM image of TiO2 hollow fibers that were obtained by calcining the composite nanotubes in air at 500 °C. (d) SEM image of a uniaxially aligned array of anatase hollow fibers that were collected across the gap between a pair of electrodes.60 | ||
3.4. Formation of 2D titanate/titania nanosheets
As mentioned above, titanate nanosheet is the intermediate during the hydrothermal process for the formation of the titanate nanotube and nanowire. Therefore, by precise control of the experimental condition, we can obtain the nanosheet morphology of the titanate materials. For example, titanate nanosheet was obtained when NaOH concentration is decreased to 5–8 M at 140 °C for 48 h under hydrothermal conditions.63 This special structure could also be obtained by organic-stabilizer-free synthesis,64 continuous hydrothermal route (flow 450 °C superheated water through a crystallizing medium),65 room temperature synthesis,66 and ionic liquid synthesis approach.67 For example, the layered titanate nanosheets (LTNSs) and carbon-supported stacked TiO2 nanosheets (CTNS) obtained from the thermal treatment of LTNSs are obtained by ionic liquid synthesis approach via the scheme in Fig. 16.67 The typical morphology of LTNSs and CTNS is shown in Fig. 16. The LTNSs have a lateral size of 20–50 nm, and layered interlamellar spacing is around ∼1 nm. The disc-like nanostructure of CTNS obtained after 350 °C annealing for 2 h is clearly evident in its high-resolution TEM image showing that several ultrathin nanosheets stack together.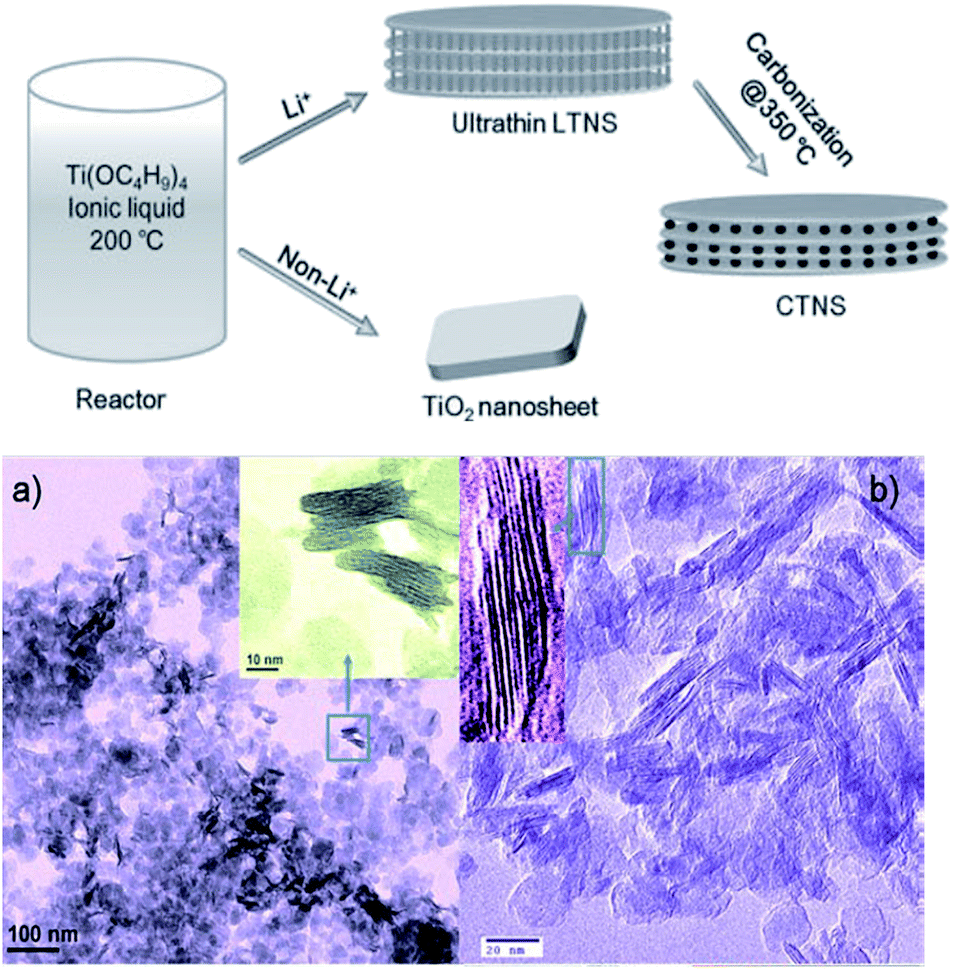 | ||
| Fig. 16 The formation scheme of layered titanate nanosheets (LTNSs) and carbon-supported stacked TiO2 nanosheets (CTNS) with a sandwich-like multilamellar structure; TEM images of the (a) as-synthesized LTNSs and the (b) CTNSs obtained after annealing at 350 °C for 2 h.67 | ||
On the other hand, the single graphene-like layered titania sheet can be prepared by delaminating a lepidocrocite-type layered protonic titanate.67,68 In this approach, TiO2 and Cs2CO3 are calcined first to form the Cs–titante. Then the Cs+ is ion exchanged by H+, and finally the titanate nanosheet is exfoliated in the presence of tetrabutylammonium hydroxide (TBAOH). Since the titania sheets possess a negative charge, the positive charge of the metal and metal oxide nanoparticles can be re-assembled with the titania sheet to form the porous nanohybrids.67,69–73 An example is shown in Fig. 17, the exfoliation-precipitation method enables the construction of a new porous material through the exfoliation of the titanate sheets and through precipitation with MgO fine particles.35 Autoclave is not required for this synthesis route, and ambient condition is sufficient for most of the stages. Nevertheless, this process suffers from some drawbacks, e.g. the need for multistage processing and the overall long processing time.
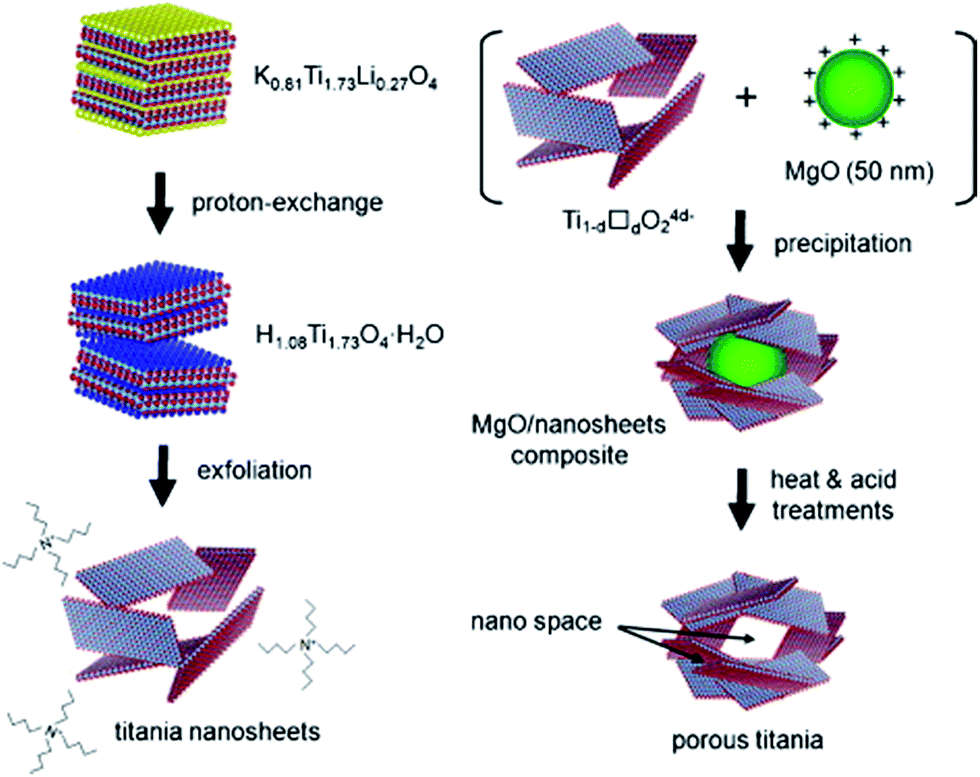 | ||
| Fig. 17 The synthesis route of titanate nanosheets by exfoliation method.35 | ||
3.5. Preparation of 3D titanate hierarchical materials
The synthesis of 1D and 2D nano-titanate has been widely investigated and well developed. However, multi-scale 3D hierarchically shaped titanate remained a challenge for many years and the synthesis is usually not straightforward. For examples, Cao et al.74 synthesized titanate flowers via a template-assisted approach through hydrothermally reacting NaOH with the mixture of TiO2 and Zn(NO3)2·6H2O, followed by the removal of ZnO flower-like nanorods framework. Yoshikawa et al.75 reported a two-step method combining hydrolysis of titanium tetraisopropoxide with hydrothermal treatment of amorphous TiO2 spheres to prepare spherical titanate nanosheet. Imai's group76 systematically explored various kinds of hierarchical ammonium titanate nanosheet morphologies grown in the ager gel medium by a bottom-up route. Mao et al.77 demonstrated the growth of micrometer scale sea-urchin-like structures by the oxidizing H2O2 assisted-hydrothermal method in autoclave at different temperatures and durations, and they proposed that a two-stage (“growth-then-assembly”) growth mechanism for the formation of titanate spheres. The self-assembled titanate nanosheets can also be synthesized via chimie-douce method by refluxing TiO2 powder in 15 M NaOH at 423 K for more than 24 h.78 Xie's group used the Kirkendall effect to construct 3D hollow titanate tubular hierarchical structures via treating TiCl4 precursor in other alkaline solutions (e.g., ethylenediamine) at 225 °C for 12 h.79 Moreover, Zhao's group reported a hydrothermal etching assisted crystallization route to synthesize Fe3O4@titanate yolk–shell microspheres with ultrathin nanosheets-assembled double-shell structure (Fig. 18).80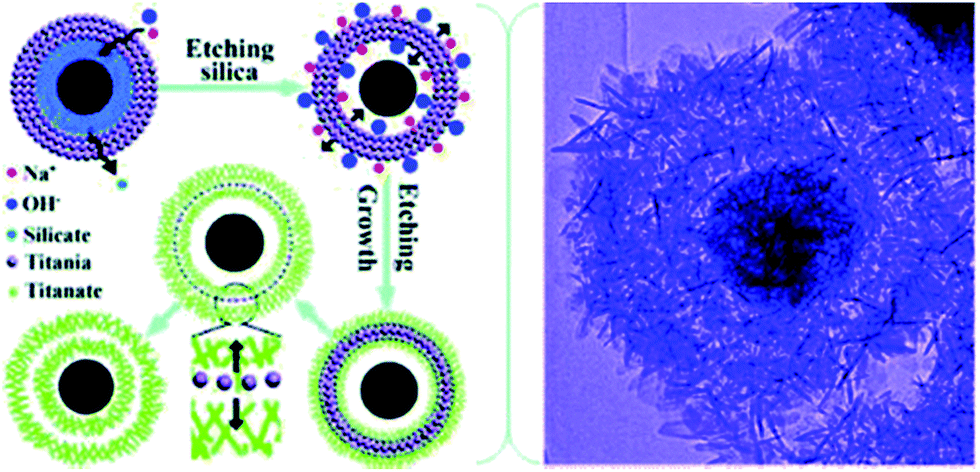 | ||
| Fig. 18 Formation scheme and TEM image of the hierarchical titanate microspheres by hydrothermal method.80 | ||
Inspired by these works, we developed a novel one-pot approach to produce highly porous hierarchical 3D titanate micro-spherulite (TMS) particles (Fig. 19) via a simple, high throughput method employing simultaneously electrochemical anodization and spark discharge of the anodized oxide layer into a reactive solution by carefully adjusting the applied electrical spark parameters in a traditional ambience setup.81,82 Electrochemical spark discharge spallation (ESDS) process is led by the concurrent anodic reaction oxidation and the electrical breakdown of the formed oxide layer into the electrolyte in precipitate form. The spallation is driven by a continuous spark discharge that simultaneously heat up the solution. The TiO2 precipitates then react immediately with the heated NaOH solution to form the TMS.
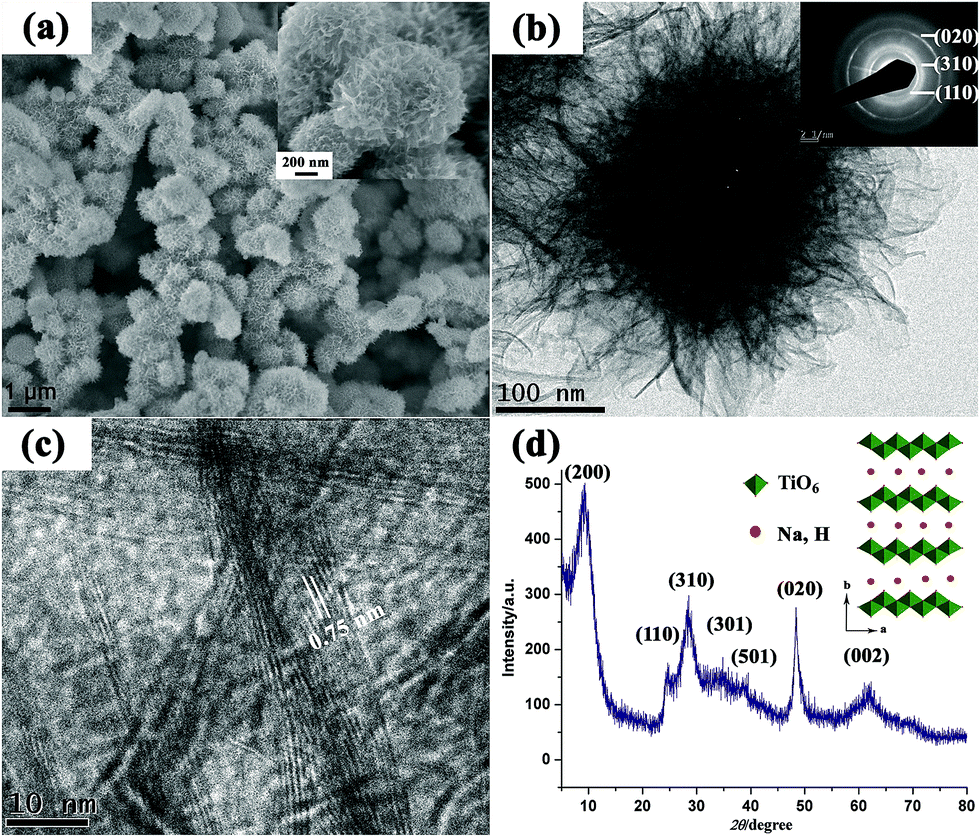 | ||
| Fig. 19 (a) FESEM image, (b) TEM image, (c) high resolution TEM image, (d) XRD pattern of the fabricated TMS. The TMS powder is collected after anodization of Ti foils in 10 M NaOH aqueous solution for 20 min. Inset shows the crystal structure for lepidocrocite titanates.81 | ||
4. Application of titanate/titania nanomaterials
Regarding this special crystal structure with layered sheets, significant attention has been paid in the field of photon-mediated water decomposition,83 photocatalysis,84 fuel cell electrolytes,85 adsorbent,86 lithium ion batteries (LIBs),87 dye-sensitized solar cell (DSSC) and wettability control related applications owing to their excellent ion-exchange/intercalation activities, as well as adsorption/photocatalytic properties. In the following sections, the titanate and titania crystal structure and surface property related applications will be discussed in detail, which are summarized in Fig. 20. | ||
| Fig. 20 Titanate and titania nanomaterials for environmental and energy applications. | ||
4.1. Adsorbent application
Adsorption is an important process in wastewater treatment, because it does not only encompass the physical removal of pollutants from water, but also is a crucial step in photocatalysis.31,88–90 During this process, pollutant molecules are adsorbed to the surface of the adsorbent. To understand the adsorption mechanisms governing titania and titanate nanostructures, the equilibrium adsorption isotherms can be fitted to two widely-used models: the Langmuir and the Freundlich equations:
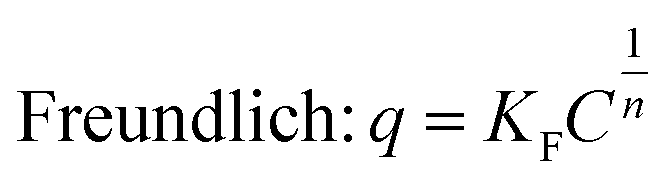 | (3) |
 | (4) |
 | (5) |
To investigate the rate at which the pollutant is adsorbed on the nanostructures, it is useful to employ the pseudo-first-order kinetic model and the pseudo-second-order kinetic model. The pseudo-first-order kinetic model can be expressed in its integral form as:
|
ln(Qe1 − Qt) = lnQe1 − k1t
| (6) |
Similarly the pseudo-second-order kinetic model can be expressed in its integral form as:
 | (7) |
In view of the crystal structure of titanate materials, sodium (Na+) or proton (H+) occupy the inter-layer cavities between the TiO6 octahedral sheets. The cations between the layers are exchangeable and titanate layer structure is stable during the ion-exchange process. For example, the interlayer distance of multilayered titanate nanotubes are not significantly altered when alkaline ions (Li+, Na+, K+, Rb+, Cs+)91 or some transition-metal ions (Cu2+, Co2+, Cd2+, Zn2+, Ni2+)92 are intercalated between the layers, demonstrating the rigidity of the titanate lattice towards ion-exchange. For the specific case of sodium ions, the maximum ion-exchange capacity, defined as the ratio between the atomic percent of sodium and titanium in the nanotubes (Na/Ti ratio), was reported to vary between 0.67 (ref. 93) and 1.1.94 The ion-exchange capacity is crystal structure dependent. For example, in an idealized crystal structure of trititanate,34 complete ion exchange of protons in the titanate to sodium ions will give a theoretical value of 0.67 according to the reaction:
| xMn+ + H2Ti3O7 → MxH2−xTi3O7x + xH+ | (8) |
Very recently, 1D titanate nanowires95,96 and nanotubes86 were shown to have excellent adsorption ability for the removal of organic molecules and radioactive toxic metal ions. According to Zhu's work,95,96 the radioactive cations (Sr2+ and Ba2+) after the ion exchange were trapped in the titanate nanofibers permanently due to the structural change, as shown in Fig. 21. This capability of trapping radioactive cations permanently allows us to isolate them from contaminated water. Thus, the titanate nanowire adsorbents do not have the issue of disposal concern regarding the risk of secondary contamination, in which the adsorbed cations are released from the adsorbents. The titanate nanotubes uptake of the inorganic arsenic (As) is efficient in terms of rate and effective in terms of capacity. With the aid of NaOH solution, the adsorbed arsenic in the titanate is reported to be able to desorb quickly.86 The reversible ion-exchange process is mainly dependent to the metal ion concentration, which drives the ion-exchange direction when the diameter of metal ion is small enough to insert into the layered distance of titanate structures. Moreover, the synthesized titanate nanotube showed an excellent adsorption properties to the different organic dyes, including basic green 5, basic violet 10 and methylene blue through an ion-exchange mechanism or electrostatic force between the dye and the titanate material at certain conditions.97 According to the crystal structure of titanate materials, alkali metal or proton ions (H+) in the TiO6 octahedral layers are exchangeable and the titanate layer structure is stable during ion-exchange process. Therefore, the different valence of metal ions (Co2+, Ni2+, Cu2+, Cd2+, Fe2+, Cu2+, Asn+, Pb2+, etc.)37,92,98–100 between the layers in the titanate materials can be exchanged with alkali metal or proton ions (H+, Li+, Na+, K+, Rb+, Cs+).91
 | ||
| Fig. 21 Structure of titanate nanowires made for removing radioactive ions from water.95 | ||
The adsorption mechanisms and kinetics are mainly dependent on the phases of TiO2 and titanate nanostructures and the type of pollutants. It has been widely reported that the adsorption of oxalic acid on Degussa P25 follows Langmuir model.88,101,102 Similarly, the adsorption mechanisms of various titanate nanostructures also follow closely to the Langmuir model.32,86,101,103–106 Lim et al. synthesized sodium titanate nanobelts and nanotubes via hydrothermal synthesis using TiO2 and TiS2 as precursors respectively, and discovered that while both nanostructures follow the Langmuir model in the adsorption of methylene blue (MB) (Fig. 22a), the TiS2-derived nanobelts exhibit much higher adsorption capacity (312.50 mg g−1) than the TiO2-derived nanotubes (256.41 mg g−1).42 The dual phase anatase/titanate nanoparticles reported by Cheng et al.32 also follow the Langmuir model in the adsorption of MB with a capacity of 162.19 mg g−1. It was also reported that the removal of MB by anatase-covered titanate nanotubes follow the Langmuir model with a capacity of 199.6 mg g−1, and that the removal of methyl orange is mostly attributed to photodegradation.89 Titanate nanotubes were also employed in the adsorption of heavy metals, such as thorium,103 chromium,104 arsenic (Fig. 22b)86 and lead.81 While the adsorption of both thorium and chromium follow the Langmuir model,103,104 interestingly, the adsorption of arsenic by protonated titanate nanotubes synthesized via hydrothermal synthesis fits very well with both the Langmuir and Freundlich models.86 The morphology dependence of titanate nanostructures (nanotubes, nanowires and nanorods) on the adsorption of formic acid was also investigated, and it was shown that the adsorption capacity of the titanate nanotubes calcined at 400 °C exhibits the highest capacity following the Langmuir adsorption model.105
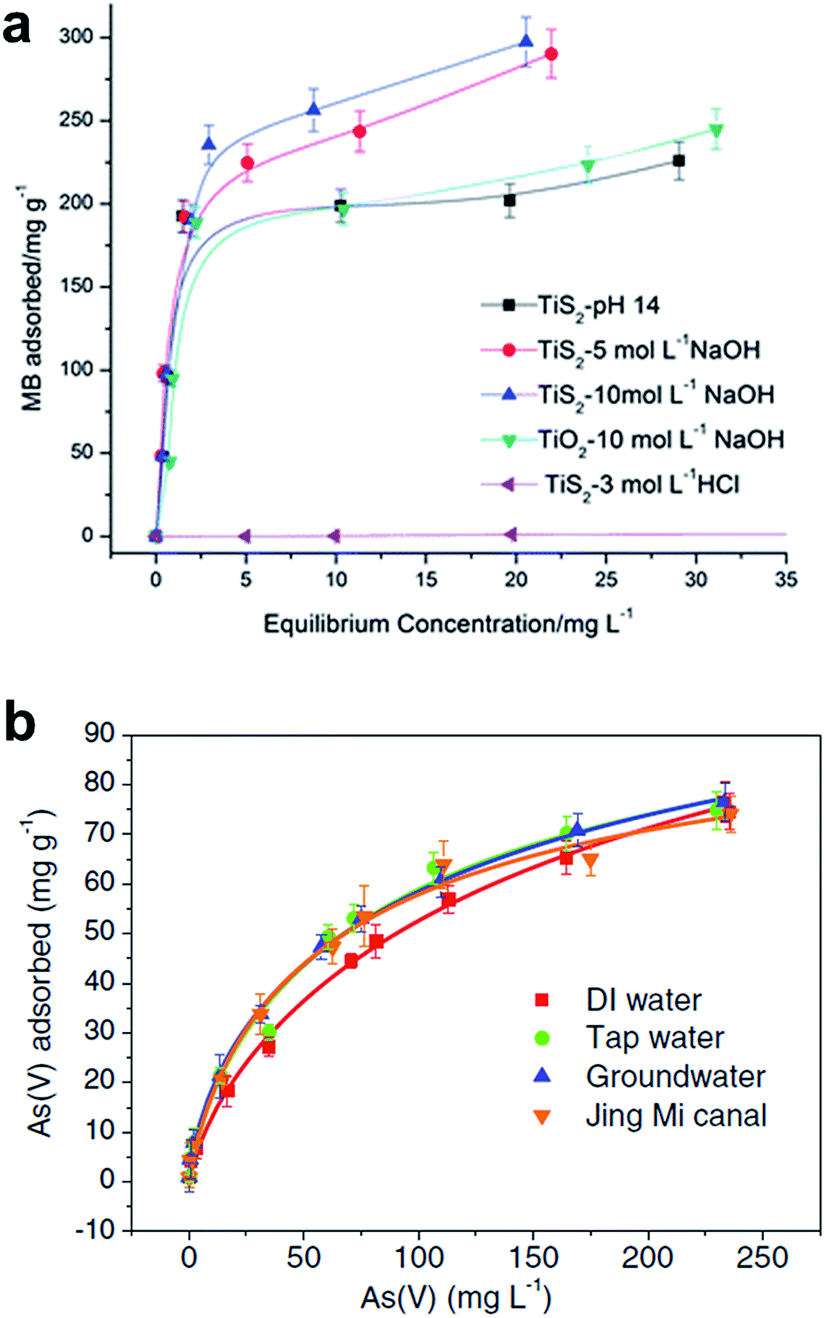 | ||
| Fig. 22 Adsorption isotherms of various titania and titanate nano-structures in MB. The y-axis displays the amount of MB adsorbed by the nanostructures, the x-axis displays the equilibrium concentration of MB.42 Langmuir isotherms for (a) As(V), and (b) As(III) adsorption on titanate nanotube in several real water samples (pH 7.0, 1.0 g L−1).86 | ||
It is useful to carry out adsorption kinetics tests to investigate the rate of adsorption of the pollutant by the various titania and titanate nanostructures. It has been observed the MB blue concentration decreased rapidly in the first 30 minutes for various titanate nanostructures, including sodium titanate nanoflowers107 and Ag–AgCl anchored on titanate nanotubes,108 which is attributed to the strong electrostatic attraction forces between MB and the negatively-charge titanate nanostructures.109,110 For example, the Ag–AgX–titanate nanotubes hybrid (Ag–AgCl–TNT) with a high surface area can synergistically adsorb and degrade methylene blue (MB) under visible light, viz., the titanate nanotubes adsorb MB onto their surfaces, which aids the degradation of MB by Ag–AgX nanoparticles (Fig. 23a–e). The excellent degradation performance of Ag–AgX under visible light is originated from its localized surface plasmonic resonance effect. The kinetics of adsorption of MB appears to be independent of morphology, as adsorption of MB by hydrothermally-synthesized sodium titanate nanobelts,42 titanate nanotubes106 and sodium titanate nanoflowers (Fig. 23f and g)107 all follow the pseudo-second-order kinetic model. In the case of heavy metals, it was reported that the adsorption of thorium (Th) by TiO2–titanate nanotubes follows the pseudo-second-order kinetic model.103 For the photocatalytic degradation of chromium (Cr) with titanate nanotubes, the synergistic process of chromium removal was found dependent on pH: adsorption of Cr(III) is promoted when pH < 5, while pH > 5 promotes Cr(IV) reduction on TiO2.104 Similarly, the adsorption and photocatalytic reaction of basic violet 10 (BV-10) dye on Co-doped titanate nanotubes also match well with the pseudo-second-order kinetic model, with the adsorption constant k2 being one order of magnitude higher than the photocatalytic rate constant. This implies that the photocatalysis of BV-10 dye is the rate-determining step.111
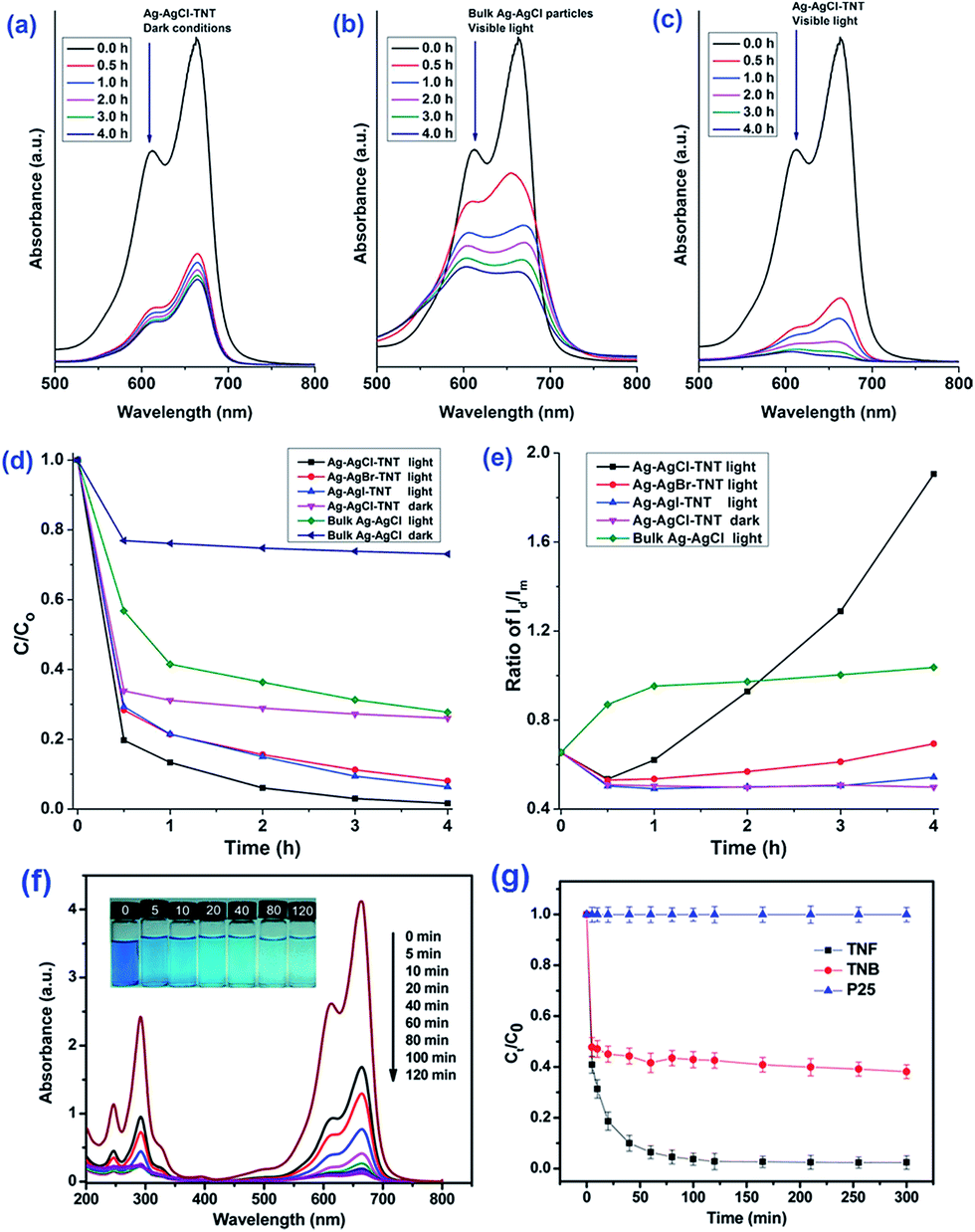 | ||
| Fig. 23 Methylene blue (MB) aqueous solution absorption spectra after treatment using (a) Ag–AgCl–TNT under dark conditions, (b) bulk Ag–AgCl particles under visible light, and (c) Ag–AgCl–TNT under visible light for different durations. (d and e) Adsorption and visible light degradation performance of different silver/silver halide based photocatalysts/conditions for MB removal.108 (f) Absorption spectra of MB solution (20 mg L−1, 50 mL) in the presence of sodium titanate nanoflowers (20 mg) under dark conditions. Inset represents the photograph of MB solutions at varying times. (g) Adsorption of MB (20 mg L−1, 50 mL) by sodium titanate nanoflowers, sodium titanate nanobelts and P25. C0 is the initial concentration of the MB solution, and C is the concentration of that at different intervals during the adsorption. Error bars show standard deviations from two independent experiments, while each experiment was performed in triplicate and the average of concentrations was taken.107 | ||
4.2. Photocatalytic application
The mechanism of photocatalytic degradation (Fig. 24) of organic compounds is similar to the TiO2-based photocatalysts. When the ultraviolet (UV) radiation photons from sunlight or illuminated light source are absorbed by the photocatalyst, it will produce electron–hole pairs. The electrons initially rested in the valence band of titanium dioxide are excited by the photon energy, and are promoted into the vacant conduction band of titanium dioxide. The resultant positive holes in the conduction band of titanium dioxide can then attack water molecule to generate hydroxyl radical (OH˙), while the negative-electrons in the conduction band can attack oxygen molecule to generate peroxide anion. The generation of surface OH˙ groups and peroxide anion are required for photocatalytic degradation of organic compounds.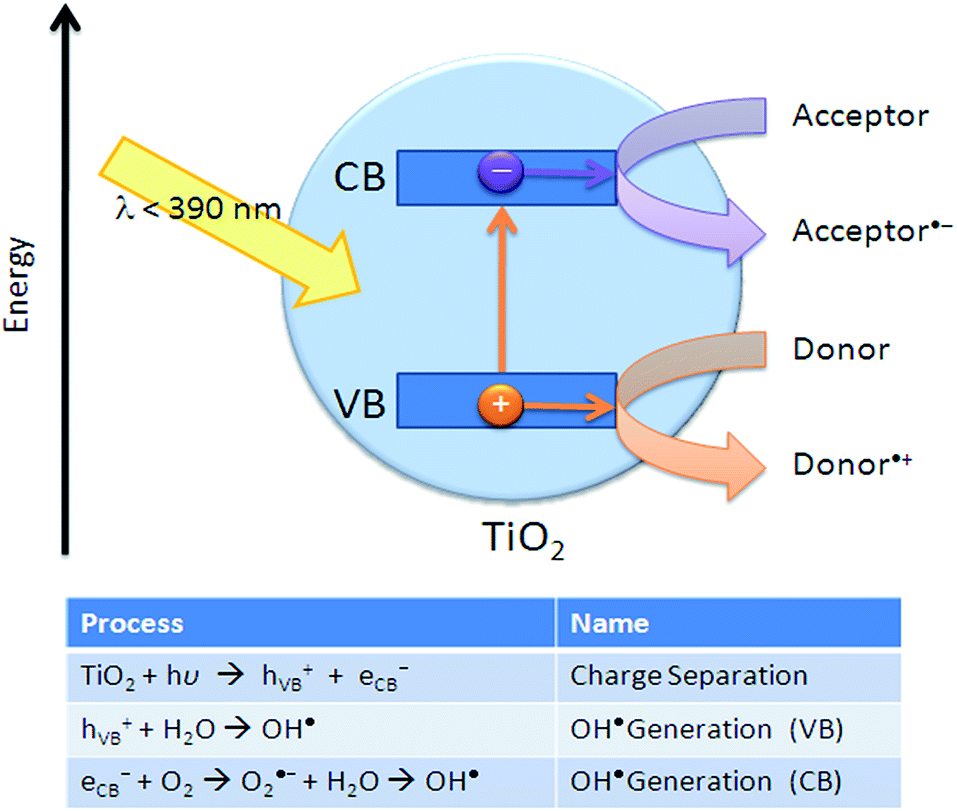 | ||
| Fig. 24 Principles of oxidative decomposition of photocatalysts.112 | ||
Titanate materials are semiconductors with a bandgap of 3.4–3.7 eV. The area of photocatalysis is one important application that these titanate materials are suited for, but the literature tackling this aspect of application has been limited. Uncertainties exist over the photocatalytic properties of the titanate materials. For example, a few researchers insisted that enough potentials are possessed by the as-prepared sodium- or hydrogen-titanates nanotubes towards the degradation of the organic pollutants.113–117 Recent work indicated that the photocatalytic activity of as-prepared titanate materials (nanotubes, nanowires, nanosheet) was not as good as the commercial standard P25 catalyst when they were tested for oxidation of NH3 and organic dyes118,119 in aqueous suspensions. Either sodium impurities in as-prepared titanate or their moderate crystallinity have been proposed to be the cause of the observed low activity. It is known that recombination centered created by Na+ impurities can lower the photocatalytic activity of TiO2.120 After alkaline hydrothermal reaction, the amount of sodium ions being intercalated into the titanate crystal is extremely high; the photocatalytic activity of the as-prepared titanates is significantly affected. The negative correlation has been confirmed recently between sodium content in the titanate and their photocatalytic dye oxidation activity.121,122 Luminescence quenching, which removes sodium ions in the structure, was observed after the protonation of titanate nanotubes.123,124 On the other hand, some researchers hold the opinion that neither sodium nor hydrogen titanate nanotubes are efficient photocatalyst due to their intrinsic inertness for photocatalytic reactions.125–128 Researches have shown that no improvement in photocatalytic activity was observed when acid was used to ion-exchange the as-prepared titanate samples. However, calcination was found to have led to apparent increase in photocatalytic activity for these titanate nanotubes. Further complication arises due to the scatter on the reported photocatalytic activity comparison experiment. For example, a 3 times better photocatalytic activity than commercial Degussa P25 was reported by Yu et al.127 on the calcined samples. However, Zhang et al.122,126 claimed that, despite the large range of calcination temperatures they have tried, the resultant photocatalytic activity of the nanotubes samples are still inferior to the commercially available powders. The inconsistency among existing reports may be due to the different preparation and test conditions from different groups.
Titanium dioxides, derived from nano-structured layered titanates, have attracted great attention during the past decade. Titanate materials provide a good platform for delicate phase- and morphological tailoring, by annealing37,129 or wet chemical reaction,115,130 to obtain various TiO2 or controlled mixed-phase nanostructures. Lots of effort has been made to improve the performance of TiO2-based materials during the past four decades, with the hope to bring it into industrial scale application. Some modest gains have been achieved so far in spite of the complexity and difficulty of the task. The main approaches could be sub-classified into three groups (as illustrated next), although more than one method might be used concurrently to modify the pristine titanium dioxide.
4.2.1.1 Optimizing the crystallinity, surface area and exposed facets. The photocatalytic activity of four titanium dioxide polymorphs has been reported before, namely anatase, rutile, brookite and TiO2(B). Their crystal structures are discussed earlier. The information, together with the band gap and density are summarized in Table 2.131,132
| Crystal structure | Crystal system | Bandgap (eV) | Density (g cm−3) |
|---|---|---|---|
| Rutile | Tetragonal | 3.00 | 4.13 |
| Brookite | Orthorhombic | 3.30 | 3.99 |
| Anatase | Tetragonal | 3.20 | 3.79 |
| TiO2(B) | Monoclinic | 3.20 | 3.64 |
Anatase and rutile TiO2 have been intensively studied for photocatalysis, in part due to their ease of preparation. Of the two common forms, anatase has a conduction band minimum sufficiently negative to reduce protons. Its photoactivity is also generally superior to that of rutile. TiO2(B), as a relative new phase,133 has attracted lots of attention as an alternative photocatalyst due to its relative open structure, although its photoactivity is generally lower than that of anatase TiO2. Furthermore, a few groups18,134,135 reported impressive photocatalytic activity of brookite TiO2, even better than known active anatase TiO2 for hydrogen production.18,135
As a special type of heterogeneous catalyst, in principle photocatalysts also benefit from similar textural effects, e.g., dispersion into smaller particles increases the specific surface area, thereby promoting faster and stronger adsorption of reactants at the interface, ultimately leading to higher rates of reaction. Problems of recombination of photo-generated charges can be mitigated by enhancing the crystallinity of the photocatalyst. Some improvements have been achieved by fabricating it in higher surface area form while maintaining a high degree of crystallization on the nano-scale with different morphologies, such as nanoparticle, nanotube, nanowire, nanoflake, etc.136–139 A hybrid TiO2 nanoflake/nanoparticle hierarchical structure formed at 500 °C was found to exhibit a higher photocatalytic activity for the degradation of organic compounds (Fig. 25a and b).138 Unfortunately, this approach is generally difficult because the above attributes are mutually antagonistic and often self-cancelling. A process that most readily increases the crystallinity through thermal treatment inexorably leads to surface area loss due to coarsening, thus dictating a practical compromise between these two factors. More recently, some success was achieved by synthesizing titanium dioxide with a high percentage of active facets.140–148 For anatase TiO2, both the theoretical prediction and experimental results show that its (001) facets are more active than the (101) facets, however, the latter normally dominates the surfaces in the equilibrium state. By fluorine-termination of anatase TiO2, the desired (001) facets can comprise up to 50% of the surface.140 Fig. 25c shows the representative scanning electron microscope image of the highly truncated bipyramidal anatase TiO2 single-crystal nanosheets. Statistically, the synthesized single-crystal nanosheets (SCNSs) have 64% the (001) facets and display superior photoreactivity (more than 5 times) compared to P25 as a benchmarking material (Fig. 25d).147 Also, the coexistence of low-index facets with a highly photoactive (001) facet in anatase TiO2 nanocrystals has been found beneficial to enhance the photocatalytic performance of TiO2 via a synergistic effect. Dufour et al.149 fabricated four different morphologies of pure anatase nanoparticles with exposed surfaces and studied the impact of the exposed surfaces on the photocatalytic efficiency. They found that the most efficient photocatalyst for rhodamine B degradation is the (101) facets TiO2, which possesses the stronger acidic surface sites. Recently, our group found that this synergistic effect can be extended from a single crystal to interconnected nanocrystals with dominating (001) and (010) facets in close contact with each other in a hierarchically structured porous particle form.150 The two faceted building blocks both belong to anatase TiO2, but they display different band gap values and band potential positions due to the dominating crystal planes. As a result, photo-generated electrons and holes can transfer from one to the other, giving rise to effective charge separation as commonly observed in a two-semiconductor heterojunction. The particles synthesized at the optimal condition showed outstanding photocatalytic hydrogen production of 364.2 μmol g−1 h−1, which is about four times as much as that of commercial P25 (96.5 μmol g−1 h−1).150 Although the promising properties of anatase TiO2 with exposing specific surfaces have been widely investigated, a clear assessment of the role of the crystal faces in photocatalytic processes is still under debate. Recently, D'Arienzo et al.151 revealed that the concentration of trapped holes (O− centers) increases with increasing the (001) surface area and photoactivity, while the amount of Ti3+ centers increases with the specific surface area of the (101) facets in vacuum conditions, and the highest value occurs for the sample with the worst photooxidative efficacy. These results suggest that (001) surfaces can be considered essentially as oxidation sites with a key role in the photoxidation, while the (101) surfaces provide reductive sites which do not directly assist the oxidative processes. Based on this work, they also demonstrated that the correlation of the reactivity on the type of crystal facets and photogenerated defects.152 The electron spin resonance spectra reveal that the amount of Ti3+ (electron traps) is parallel to the H2 evolution rate and becomes a maximum for the nearly rectangular TiO2 nanocrystals, which display the highest area of the (010) surfaces and lowest (010) area but also involve a significant fraction of the (010) facets.
 | ||
| Fig. 25 (a) SEM images of hybrid nanoflake/nanoparticle hierarchical structure and (b) its photocatalytic activity;138 (c) typical SEM images of the anatase TiO2 single-crystal nanosheets (SCNSs) and (d) normalized fluorescence intensity per unit surface area with different photocatalysts: clean SCNSs, TiO2 SCNSs capped by F atoms (denoted as F-SCNSs), and Degussa P25 TiO2.147 (e) A correlation between the nature of the exposed surfaces, the corresponding surface acidic behavior. Photocatalytic degradation curves of the RhB solution by the anatase samples with (101) exposed facet and the P25 reference.149 | ||
4.2.1.2 Development of TiO2 based mixed-phase photocatalyst. Although the relative photocatalytic efficiency of anatase and rutile TiO2 is still under debate, it is now well-established that mixed-phase photocatalysts show enhanced performance as compared to either single phase. The success of Degussa P25 TiO2, the benchmark widely used for comparison of other photocatalysts, is a prime example though it is not purposely designed for such benefit. Commercial Degussa P25 is made by the “aerosil process”, yielding individual particles consisting of roughly 20% rutile and 80% anatase.153 Its high performance is generally attributed to the abundance of “rectifying” phase junctions that facilitate unidirectional (and opposite) flow of free carriers, thereby facilitating charge separation.154 However, the nature of the atomic level contact between anatase and rutile is still not well understood,155 and it has been demonstrated that the distribution of phases can be further improved for photocatalytic action by controlled annealing.156 Based on the above principle, anatase and TiO2(B) core–shell structured nanofibers have been synthesized by hydrothermal treatment of titanate nanobelts, and their efficiencies were found to be comparable to that of commercial Degussa P25 (Fig. 26).129,157 Highly crystalline pure brookite and two-phase anatase/brookite TiO2 nanostructures were also successfully synthesized via a simple hydrothermal method with titanium sulfide as the precursors in sodium hydroxide solutions, and the similar synergy of two phase system has been confirmed.18
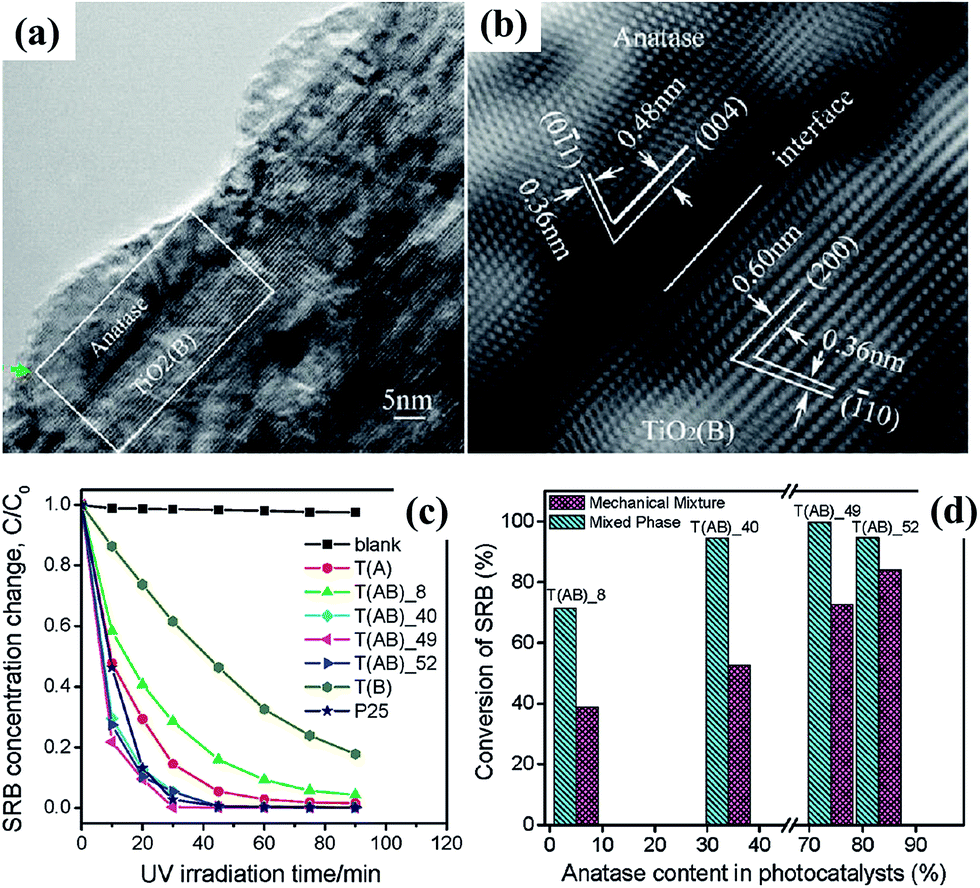 | ||
| Fig. 26 (a and b) HRTEM images of the joint of anatase nanocrystal and TiO2(B) core in a single TiO2 fiber; (c) photocatalytic decomposition of sulphorhodamine dye with different fibril TiO2 photocatalysts under UV irradiation; (d) comparison of conversion rate for the decomposition of SRB with mechanically mixed TiO2 photocatalysts.129 | ||
4.2.2.1 Bulk doping of TiO2. The optical response of any material is determined by its intrinsic electronic structure, which is a strong function of chemical composition, crystal structure, and, to some extent, physical dimension (quantum size effects). The chemical composition of TiO2 can be altered either by replacing the cation (titanium) or the anion (oxygen), referred to here as metal and non-metal doping, respectively.161–163 Historically, the most popular approach has been explored by doping with color centers, such as transition metal cations, but with mixed results. When TiO2 is doped with altervalent transition metal ions, such as CoII, NiII, CuII, FeII/III, CrIII, VIII, MnIV, NbV, MoV/VI, an electron occupied level will be formed and the electrons are localized around each dopant.131 As the dopant concentration increases, the localized level shifts to lower energy and can even reach the top of valence band for CoII.162 Electrons can be excited from this defect state to the conduction band by photons with a lower energy, i.e., in the visible range. Band gap narrowing of TiO2 can also be achieved by non-metal doping, using anions such as N, C, B, F, P, S and I.131,163–168 N and C doping are the two most investigated cases. In the pioneering work by Asahi et al.,163 substitutional doping of N (for O) was proposed as the most effective band gap narrowing due to mixing of p states between N and O. However, accumulating evidence suggests that N-doping introduces mid-gap levels slightly above the O 2p valence band.131 For C–TiO2, the dopant introduces deep states in the gap.166,167 The intrinsic defects of TiO2, such as reduced Ti species and oxygen vacancies, also lead to the formation of localized states which may merge to form sub-band gap level, thus creating a lower energy excitation pathway.131,169
Titanate, as an alternative starting material with relative open structure, is ideal for high concentration homogenous doping. In contrast with most reported nitrogen-doped titanium dioxide photocatalysts with some localized states in the intrinsic band gap and small visible light absorption shoulders induced by inhomogeneous nitrogen doping near the particle surface, the homogeneous substitution of O by N in all particles of layered titanates exhibited extraordinary band-to-band excitation in visible-light ranging up to blue light (Fig. 27).161 The actual photocatalytic efficiency of doped TiO2-based material is largely dependent on dopant concentration and homogeneity, which are highly dependent on the preparation method. Although doping with either metal or nonmetal elements definitely confers visible light absorption and associated photo-activity, the overall performance under UV-vis irradiation may even fall below that of pristine TiO2 due to deterioration of crystallinity and/or creation of particular defect sites that tend to promote charge recombination.
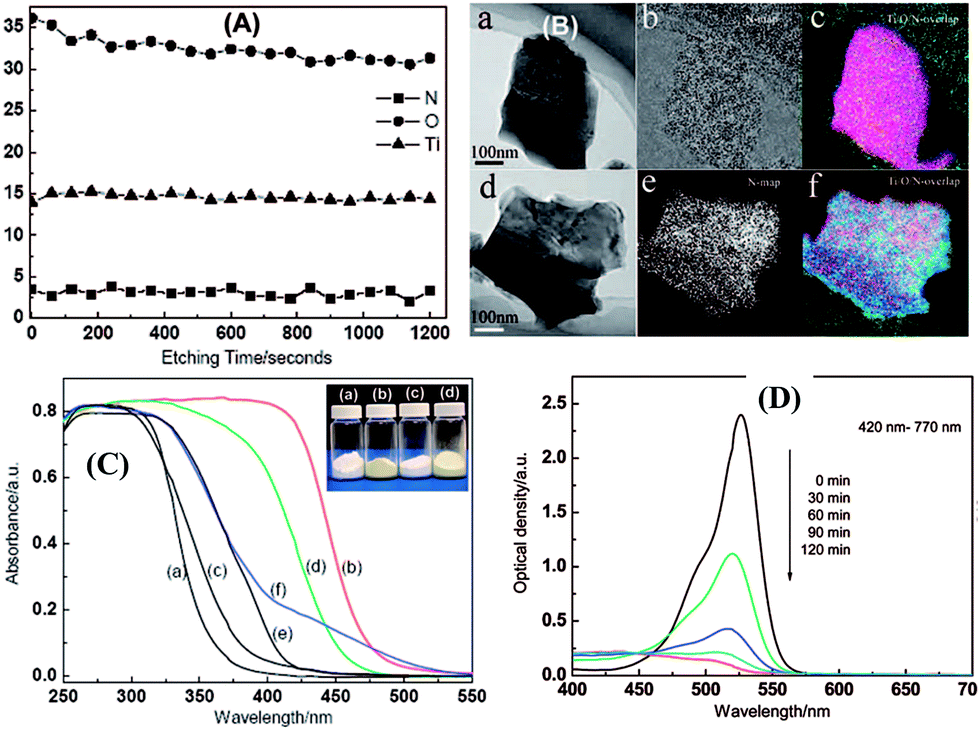 | ||
| Fig. 27 (A) XPS depth profiles of the elements of Ti, O and N in the H0.68Ti1.83O4−xNx upon Ar+ sputtering. (B) Energy-filtered TEM images of (a and d) general morphology, (b and e) N map, and (c and f) Ti/O/N overlapped maps of (a–c) Cs0.68Ti1.83O4 and (d–f) Cs0.68Ti1.83O4−xNx, respectively; (C) UV-visible light absorption spectra, (D) the photocatalytic activity of the H0.68Ti1.83O4−xNx photocatalyst.161 | ||
4.2.2.2 Surface modification with melon and carbon nitride. While the evidence has been steadily accumulated that visible light activity in TiO2 results from introducing N, bulk-doping is a complex process, and the coloration due to the sole presence of N is not easy to prove because oxygen vacancies are created simultaneously to maintain charge balance. Most of the preparation methods use an organic compound as N-source in a wet chemical process, usually followed by calcination at mild temperatures. The choice of nitrogen source, as well as the preparation conditions, have a large influence on photocatalytic activity in the resulting product, possibly due to the presence of diverse nitrogen species, such as oxidic (NOx), nitridic or amidic (NHs). Recent works focusing on cheaper wet-chemical methods, typically based on urea, triethylamine, etc., as nitrogen source, have seen some success even after mild calcination of the impregnated material, where substantial bulk (ionic) diffusion is unlikely. Consequently, some researchers even doubt that doped- or surface states of N exist as such170,171 and the term “N-modification” is generally preferred. There is now strong evidence that “melon-like” surface layers are obtained170,171 when starting from urea. The most commonly used nitrogen sources are urea and its derivatives. A careful study by Mitoraj and Kisch170–172 based on urea has clearly proved that a condensed polymeric layer of melon is formed on the TiO2 surface, and it is the chromophore that is responsible for visible light activity. As this surface-overlayer-based system is quite distinct from the (bulk) doped type, they state that it is best classified as “melon-modified” TiO2.
The formation of melon from urea is a condensation process involving the surface hydroxyl groups of the substrate. It can be conveniently divided into three stages:170,171 (1) thermal decomposition of urea into isocyanic acid and ammonia at 320–400 °C (eqn (9)); (2) cyanamide, and eventually melamine, formation by the reaction of iscocyanic acid with surface OH groups of TiO2 (eqn (10) and (11)). The balance of these reactions is melamine formation from urea in the presence of TiO2 at 400 °C (eqn (12)); (3) melamine then undergoes polycondensation to form melem, melam and finally melon (Fig. 28). The (yellow) melon is believed to be the principal structure conferring visible light sensitization.
 | (9) |
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ti–OH + O Ti–OH + O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–H → Ti–NH2 + CO2 CN–H → Ti–NH2 + CO2
| (10) |
|
Ti–NH2 + H–O–CN → Ti–OH + H2N–CN
| (11) |
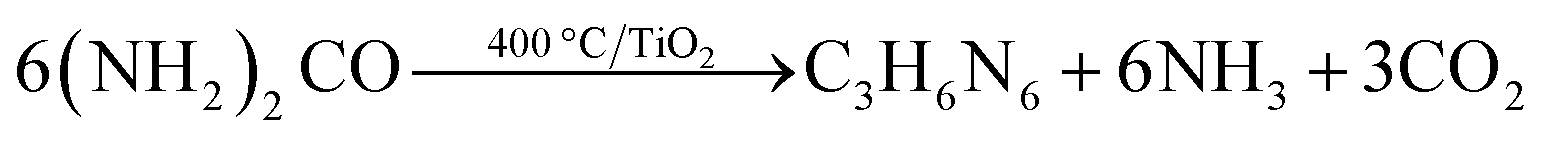 | (12) |
 | ||
| Fig. 28 Scheme illustrating polycondensation of melamine to melam, melem and melon.171 | ||
As compared to titanium dioxide, the layered titanate nanotubes/nanobelts offer a practical advantage by promoting the build-up of melon from urea. This is believed to be linked to the higher density coverage of titanates by surface OH groups and their Brønsted acidic properties, which promote polymerization.173 A dual-phase material composed of hydrated titanate and anatase (TiO2) synthesized through a low-temperature one-pot process in the presence of trimethylamine (TEA) as the N-source also exhibited visible light photocatalytic activity that can be attributed to a polyunsaturated hydrocarbonaceous overlayer.174 Similar observations were discovered for the so-called “C-doped” titanium dioxide. Kisch et al.175 demonstrated that some organic species containing carboxylic groups can be extracted from the surface of “C-doped” samples by heating them in the alkaline solution, thus the visible light activity of many previously reported so-called “C-doped” titania powder probably does not originate from the presence of lattice carbon atoms or oxygen defects, but from the surface species.
4.2.2.3 Metal and narrow band gap semiconductor sensitization. Another alternative approach to extend the light absorption to visible range is to create TiO2-based composites with colored (narrow band gap) semiconductors such as CdS, CdSe, etc.,176–179 where a synergy is evident due to directional inter-phase charge transfer analogous to mixed-phase TiO2. For examples, Zhang et al.176 demonstrated that CdS–titanate spheres, termed as hierarchical echinus-like titanate spheres (HETSs), showed effective photocatalytic activity for visible-light-driven H2 (Fig. 29). This work confirms that there is fast electron injection (∼0.32 ns) between these two kinds of semiconductors (CdS and titanate). This process where the photoinduced electrons on the CdS quantum dot rapidly localizes to the titanate material significantly enhanced the charge separation, therefore promoting the H2 yielding compared to the pure CdS sample. On the other hand, noble metal nanoparticles have been widely used as surface modifiers for TiO2-based photocatalysts. These enhance charge separation by acting as “electron sinks”,180,181 thereby promoting efficient photo-reduction. Noble metal nanoparticles, especially Au and Ag, have the unique property of being optically excited in the visible region due to the so-called surface plasmonic resonance (SPR) effect.182–185 These materials are now under intensive investigation as potential visible sensitizers.186 In well-defined nanostructures, localized surface plasmon resonance (LSPR) can occur when confined free electrons oscillate with the incident light irradiation at the same frequency. Under LSPR excitation, plasmon can “excite” the electrons in the conduction band to produce highly energetic “hot electrons”. These “hot electrons” can escape from the plasmonic nanostructures and transfer to a semiconductor in contact, thereby forming a metal–semiconductor Schottky junction.182,183,187 The semiconductor in contact with plasmonic metal nanostructures has a great effect on the charge injection efficiency. Good alignment of the Fermi level of the plasmonic metal nanostructures with the semiconductor band favors efficient photo-generated charge carrier injection.188
 | ||
| Fig. 29 (a) TEM image of CdS–hydrogen titanate spheres; (b) UV-vis diffused reflectance spectra of pure CdS, HETSs and CdS–HETSs samples; (c) H2 evolution of pure CdS, HETSs and CdS–HETSs; (d) transient absorption (TA) decay profiles (over the first 100 ps) for the pure CdS and CdS–HETSs samples obtained with 400 nm pump/550 nm probe pulses.176 | ||
Although gold or silver nanoparticles supported on insulating oxides are reported to have photocatalytic activity under visible light,189–192 this has been attributed to artifacts, e.g., enhanced thermal catalysis caused by equilibrated local-heating effects189 and/or instantaneous heat-assisted photo-excitation of Ag electrons to higher energy states (Fig. 30a and b).190 TiO2 is by far the most frequently used semiconductor material. A series of papers have reported that Au nanoparticles supported on TiO2 surface has much more pronounced activity under visible light irradiation compared with that supported on insulator materials, and, more importantly, the action spectrum for photo-activity resembles the SPR absorption spectrum implying that injection of the plasmonic “hot electrons” from the metal nanostructures to the semiconductor in contact is indeed possible.184,193,194 Other semiconductors, such as silver halides (AgCl, AgBr, AgI), can also serve as electron acceptors,195–204 and the electron generated from the Ag nanoparticles due to the surface SPR effect is proven (Fig. 30c and d).203
 | ||
| Fig. 30 (a and b) The diagram of the band structures of the supported silver NPs and the proposed photocatalysis mechanism;190 (c) comparisons of 470 nm transient absorption kinetics of Ag nanoparticles and Ag nanoparticles coupled with AgCl nanoparticles after 400 nm excitation; (d) the proposed mechanism of photocatalytic organic dye degradation for the Ag/AgCl hybrid structures.203 | ||
4.3. Lithium-ion batteries applications
Rechargeable lithium-ion batteries (LIBs) have attracted intense attentions in recent years for their potential applications in electric vehicles.205–209 As shown in Fig. 31a, the basic operating principle of LIBs is based on Li+ shuttling reaction between the cathode and the anode. During cell discharging process, Li+ intercalates the positive materials through the electrolyte, while electrons flow through external circuits to provide electricity. During cell charging, the process is reversed. Therefore, the properties of the active Li storage materials greatly influence the performance of rechargeable LIBs. In general, electrode materials that can accommodate large number of lithium will lead to high specific capacity of the battery, while electrodes materials with open structure and short diffusion length allowing for fast ionic/electronic transfer can lead to high current density, and consequently high power density. Conventional graphite electrodes suffer from poor safety characteristics, short cycle life and poor low temperature performances. In searching for better substituents, layered titanate materials emerge as attractive alternatives owing to their open, mesoporous structures with intrinsic high safety. As discussed before, the interlayer distance of titanate nanotubes/nanowires is typically around 0.8 nm, while H+/Na+ ion are loosely distributed in the interlayer space. This makes them good candidates to provide fast diffusion channels for reversible lithium ion intercalation and de-intercalation, allowing lithium ions to be readily transported and effectively ion-exchanged. The overall lithiation reaction for titanate materials is listed in eqn (13). Factors including the number of available lithium reduction sites and the surface area determine their capacity and power characteristics collectively.| H2/3TiO7/3 + x(Li+ + e) → H2/3LixTiO7/3, (1 ≥ x ≥ 0) | (13) |
 | ||
| Fig. 31 (a) Schematic on the operating principle of traditional rechargeable LIBs;205 (b) hierarchical titanate nanostructures for lithium ion battery application.215 (c) Hydrogen titanate nanowires as high rate performance electrodes.217 | ||
So far, high specific capacity and high rate capacity, accompanied by long-term stability and high safety in operation have been achieved in the literature using titanate materials.14,210 In terms of specific capacity, earlier work by Li et al. suggested that an initial discharge capacities of 296.6 and 282 mA h g−1 at current densities of 0.3 and 0.24 A g−1 can be achieved by hydrogen titanate nanowires and nanotubes electrodes, respectively.211,212 Potassium titanate nanowires electrode displayed an higher initial discharge capacity of around 305 mA h g−1 at a current density of 0.05 mA cm−2.213 Hierarchical titanate microspheres synthesized by Xu's group exhibits even larger initial discharge capacity of 390 mA h g−1 at current densities of 0.01 A g−1, which is superior to that of the hydrogen titanate nanotube, which is 352 mA h g−1 at the same rate.214 Later, Zhang et al.215 also demonstrated that hierarchical titanate spheres (Fig. 31b) with large surface area (406.41 m2 g−1) possess a higher reversible capacity and better cycling stability compared to 1D titanate nanostructures (tubes and wires). Specifically, an initial discharge–charge capacity of 419 and 302 mA h g−1 is achieved at current rate of 50 mA g−1, even after 100 cycles, a high reversible capacity of 200 mA h g−1 can be obtained, which is superior to other titanate nanomaterials. In terms of rate capacity, Wang et al. synthesized hydrogen titanate nanowires with Ti(OC4H9)4 as the precursor, which delivers a high capacity of 282 and 194 mA h g−1 at current rate of 1 and 2 A g−1.216 The best rate performances of the hydrogen titanate based materials are reported by Zhou's group (Fig. 31c),217 where TiAl was used as the precursor and emerged into the concentrated NaOH solution for hydrothermal treatment, the obtained hydrogen nanowires (H2Ti3O7) exhibit excellent rate capacities. In particular, at current rate of 1 A g−1, a capacity of 140 mA h g−1 can be obtained after 200 cycles, even at extremely high rate of 10 and 40 A g−1, a stable capacity of 120 and 100 mA h g−1 are delivered.
Although good capacity and rate capacity have been achieved with titanate materials, the as-obtained hydrogen titanate usually contains structural water, which will react with the organic electrolyte and result in low columbic efficiency in the first cycle and decreased cycle stability. By thermal treatment of the pristine protonated titanate materials, titanate materials will lose the structural water and transform into TiO2(B) nanostructures. Compared to titanate, the nanotubular morphology and open channel structure are retained in the newly formed TiO2(B) materials with improved stability and cycle capacity. Of the most studied polymorphs,218 nanostructured TiO2(B) has the highest capacity with promising high rate capabilities. TiO2(B) is able to accommodate one Li+ per Ti, giving a capacity of 335 mA h g−1 for nanotubular and nanoparticulate TiO2(B). Pioneering works regarding the lithium-ion battery performances of the TiO2(B) electrodes are done by Bruce et al.219,220 The fabricated TiO2(B) nanowires exhibit a first cycle discharge capacity of 305 mA h g−1 at a current density of 0.5 A g−1, corresponding to x = 0.91 for LixTiO2(B). Full cell based on TiO2(B) nanowire anode coupled with LiFePO4 and LiNi0.5Mn1.5O4 cathode also demonstrated good cycling stability and rate capability.221 Similarly, TiO2(B) nanotubes also offer good capacity and cycle stability.222,223 The original method in Bruce's work involves static hydrothermal process developed by Kasuga20 to synthesize TiO2(B) materials, which limits the length of TiO2(B) nanotubes to be less than 1.0 μm. Recently, hierarchical porous TiO2(B) with thin nanosheets, which combines the superiorities of the TiO2(B) polymorph with porous structure as well as thin nanosheets, demonstrated faster insertion and extraction of Li-ion than its nanotube and nanowire counterparts (Fig. 32a).224 Tang et al. synthesized ultralong TiO2 nanotubes via a novel stirring hydrothermal technique; the electrode demonstrates excellent rate capacities and cycle life owing to intrinsic high aspect ratio and high conductivity of the long nanotubes. In particular, at rate of 25C, a capacity of 114 mA h g−1 can be delivered after 10000 cycles, maintaining 100% efficiency (Fig. 32b).10 In addition, benefited from the highly viscous nature of the nanotubes, additive-free TiO2 electrode could be fabricated and the correlation between aspect-ratio and electrochemical performances of the TiO2 materials is unveiled.54 In particular, high aspect-ratio TiO2 nanotubes deliver superior high rate capacities and exceptional long cycle life.
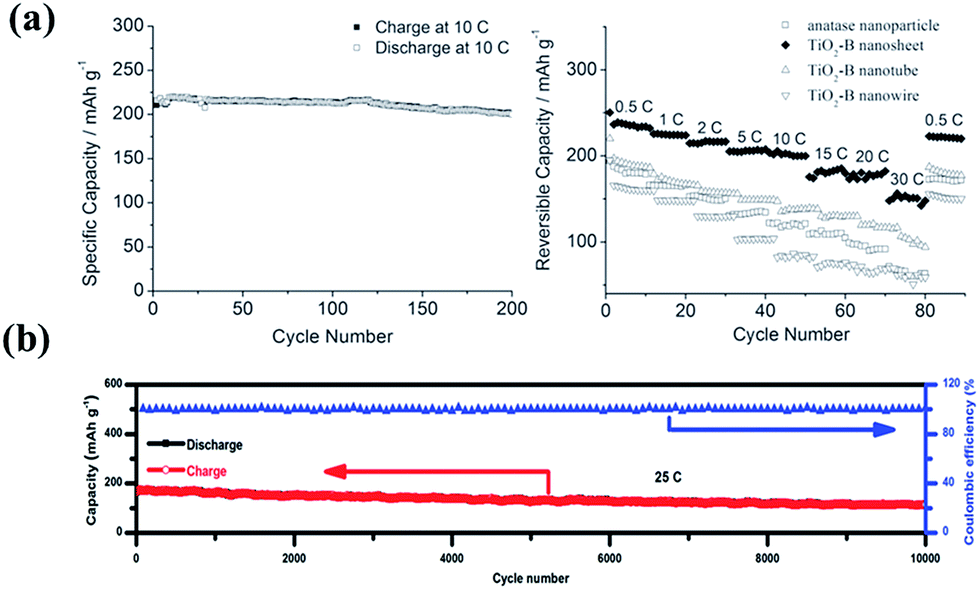 | ||
| Fig. 32 (a) Cycling performance of anatase nanoparticles, TiO2–B nanosheets, TiO2–B nanotubes, and TiO2–B nanowires at different charge–discharge rates;224 (b) long-term cycling performance of elongated TiO2(B) nanotube electrodes at a high current density of 25C.10 | ||
These results show the potential of titanate and TiO2(B) electrodes in replacing the commercial carbon negative electrodes to offer excellent rate capacity and ultralong cycle life with enhanced safety benefited from the high lithiation potential. The problems related to lithium electroplating and charge loss arising from the formation of a solid–electrolyte interface layer are also avoided.
4.4. Photovoltaic cells and electrochromic devices
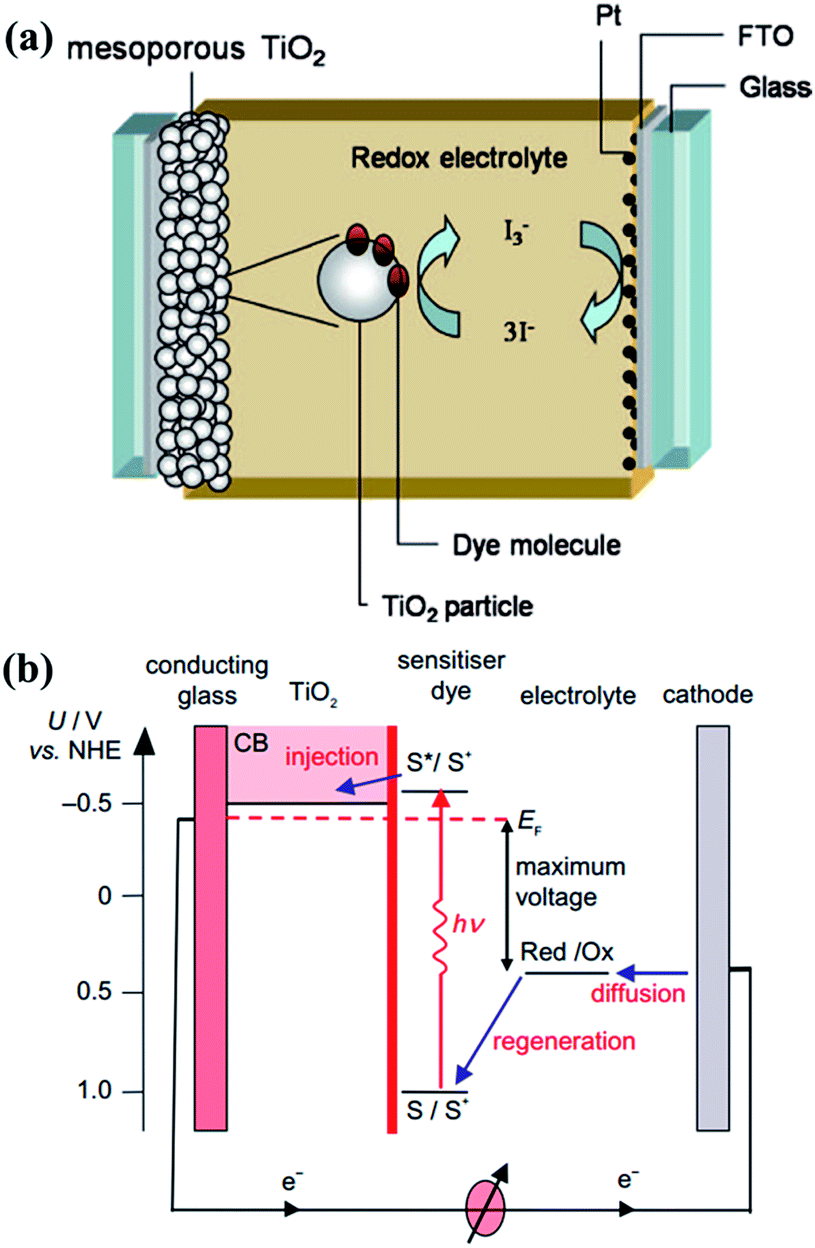 | ||
| Fig. 33 (a) Schematic overview of a dye-sensitized solar cell (DSSC) and (b) its energy level diagram. The potentials for a DSSC based on the N3 dye, TiO2, and I−/I3− redox couple are shown.231,232 | ||
At early stage of the development, porous TiO2 nanoparticle network was used as the photoanode of the DSSC,225 and promising photo-conversion efficiency was achieved. For example, Tebby et al.233 reported a UV-irradiation route towards low-temperature processing of nanoporous thin films by hydrothermal approached, and an overall energy conversion efficiency of 2.4–2.5% was obtained on 1–3 μm-thick films modified with Dyesol N3 dye. Magne et al.234 investigated the effect of TiO2 polymorphism (anatase, rutile and brookite) on DSSC properties. Their results revealed that the conductivity and diffusion coefficients followed the order of rutile < brookite < anatase, and anatase particles showed the best DSSC performance (ca. 8%). However, for TiO2 nanoparticles, the overall efficiency is limited due to electron loss during percolation through the nanoparticle network and photo-induced carrier recombination. In order to increase the overall conversion efficiency, different TiO2 structures, surface modification and morphology design have been explored to enhance the electron transfer ability. Since many excellent reviews have summarized the recent development on the DSSC with electrode structure, organic dye and redox electrolyte design and development, we only focus on the typical examples on the TiO2 nanostructure for the photoanode of DSSC applications in the current review. 1D nanostructure is an active research area for DSSC. For example, long, vertically aligned titania nanotubes235 were prepared on transparent conducting oxide glass (Fig. 34a and b) with lengths between 0.3 and 33.0 μm using a novel electrochemistry approach. Dye sensitized solar cells containing these arrays yielded a power conversion efficiency of 6.9%. Moreover, a 50 μm thick layer of TiO2-coated ZnO nanowire array coating236 (Fig. 34c) was developed for solid-state DSSC cell, which yielded an average power conversion efficiency of 5.65%. Unfortunately, these 1D arrays suffer from a greatly reduced surface area per unit volume due to their collapse and lack of nanoporosity associated with the roughness of nanometre-sized crystals.237 Much effort was then drawn towards the fabrication of hierarchical structures with high surface area. More recently, Zhao et al.238 reported a simple evaporation-driven oriented assembly method to synthesize three-dimensional open mesoporous TiO2 microspheres (Fig. 34d), which has a photoelectric conversion efficiency of up to 12.1%. Tétreault et al.239 presented a bottom-up synthesis of a high electron mobility and highly light scattering macroporous photoanode (Fig. 34e and f) for dye-sensitized solar cells, which reduces recombination and a large surface-area mesoporous anatase guest for high dye loading.
 | ||
| Fig. 34 (a) FESEM image of a 20 mm-long nanotube array and (b) digital images of transparent films of different thickness.235 (c) SEM image of a four-layer TiO2-coated ZnO nanowire array by the multistep process.236 Scale bar, 20 μm. (d) TEM images of a single ultramicrotomed, mesoporous TiO2 microsphere (inset is the model).238 (e) Schematic representation of the synthesis method for a 3D host–guest DSSC and (f) SEM of a self-assembled 3D Al/ZnO|TiO2| host–guest photoanode.239 | ||
Stemming from DSSC, perovskite solar cells based on organometal halides represent an emerging photovoltaic technology, which is selected as one of the biggest scientific breakthroughs of 2013.240,241 In 2009, a power conversion efficiency (PCE) of around 3–4% was achieved on methylammonium lead halide (CH3NH3PbI3) perovskite/nanocrystalline TiO2 based on a liquid-based DSSC structure (Fig. 35a).242 However, the stability issues (e.g., dissolution of the perovskite) of liquid-based perovskite solar cell limit their practical applications. In 2012, a long-term stable, and high efficiency (10%) perovskite solar cell was developed by substituting the solid hole conductor with a liquid electrolyte.243 Moreover, a CH3NH3PbI3/TiO2 heterojunction solar cell (Fig. 35b and c) with a p–n junction structure was reported in 2013.244 The CH3NH3PbI3 was used as a p-type semiconductor. This perovskite solar cell initially showed a PCE of 5.5% with a 500 nm-thick nanosheet TiO2 film as the n-type layer, and the higher performance approaching 11% was achieved based on this concept.241 In 2013, the PCE efficiency exceeding 15% was reported using organolead halide perovskite.245 In 2015, Seok's team246 showed that the incorporation of methylammonium lead halide perovskite (MAPbBr3) into formamidinium lead iodide (FAPbI3) stabilizes the perovskite phase of FAPbI3 and improves the power conversion efficiency of the solar cell to more than 18 percent. Most recently, a new record certified nonstabilized efficiency of 20.1% was achieved by KRICT.247 The main advantage of perovskite solar cells is their ability to perform all three basic tasks required for solar cell operation: light harvesting, charge generation and transport. In addition, their absorption coefficients can be as high as one order of magnitude greater than ruthenium complexes, and they feature long-range charge transport properties.248 Since PCE values over 20% are realistically anticipated with the use of cheap organometal halide perovskite materials, perovskite solar cells are a promising photovoltaic technology. The recent progresses on perovskite materials and perovskite solar cells are summarized in the recent reviews.241,248
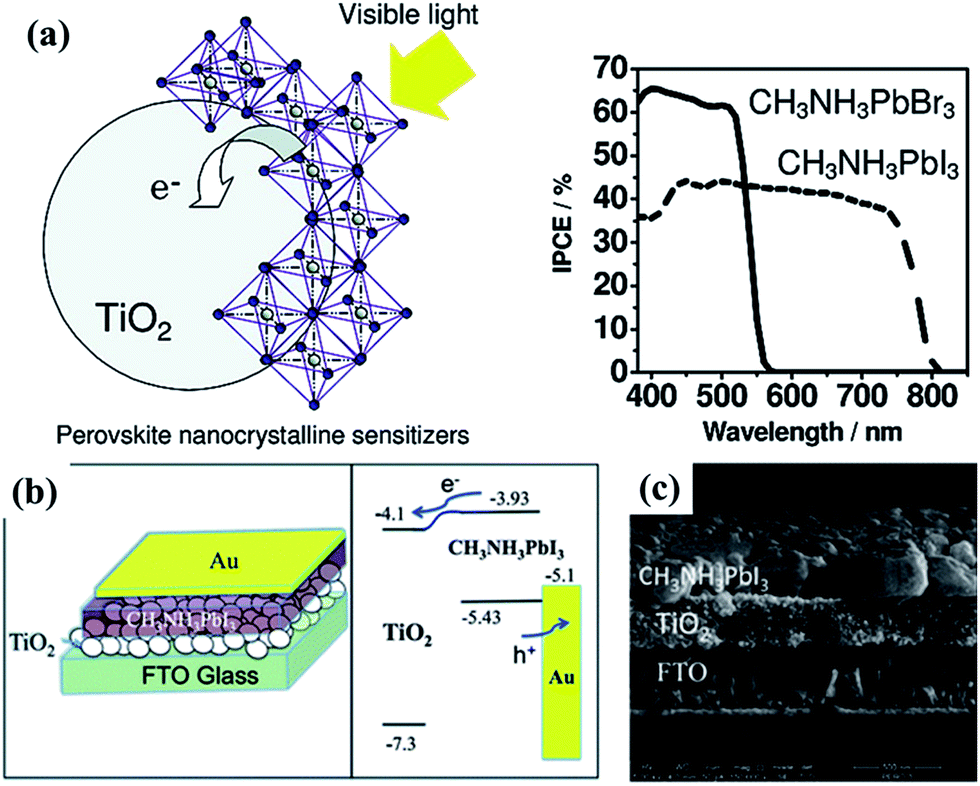 | ||
| Fig. 35 (a) Two organolead halide perovskite nanocrystals, CH3NH3PbBr3 and CH3NH3PbI3, were found to efficiently sensitize TiO2 for visible-light conversion in photoelectrochemical cells.242 (b) Schematic illustration of the CH3NH3PbI3/TiO2 heterojunction solar cell and energy level diagram of the discussed solar cell; (c) SEM cross section of the heterojunction solar cell.244 | ||
Because the specific surface area of the host materials (ion intercalation capacitance) and the defined supply of intercalating ions (electrolyte diffusion path) have great effect on the electrochromic reversibility, aligned TiO2-based nanowires and nanotubes constructed by hydrothermal and electrochemical anodizing strategies are particularly interesting in view of electrochromic devices assembling. Campet et al.251 demonstrated that TiO2 nanoparticle films constructed by a combination of hydrothermal and doctor-blade process exhibited high electrochromic responses and coloration efficiencies in a stable and environmental friendly ionic liquid electrolyte containing a lithium salt. Schmuki et al.252 reported that vertical aligned TiO2 nanotubes films formed by electrochemical anodizing directly on Ti substrate also displayed better electrochromic abilities regarding Li+ and H+ ions intercalation than nanoparticle coatings or compact oxide films. To achieve a superior optical contrast and cycle stability, researchers have also developed various strategies to create TiO2 nanotube films on transparent conductive substrate, such as ITO or FTO glass (Fig. 36).253–255 Furthermore, modification of TiO2 nanostructured films include decoration of electrochromically more active nanoparticles (e.g., WO3, NiO, V2O5, MoO3), single mixed oxide or multilayered metal oxide grown from some transition metal alloys (e.g., Ti–W, Ti–Nb, or Ti–V) were demonstrated to lead to a positive effect on the threshold responsive potentials, higher transmittance contrast, and/or longer cycling stability.250,256
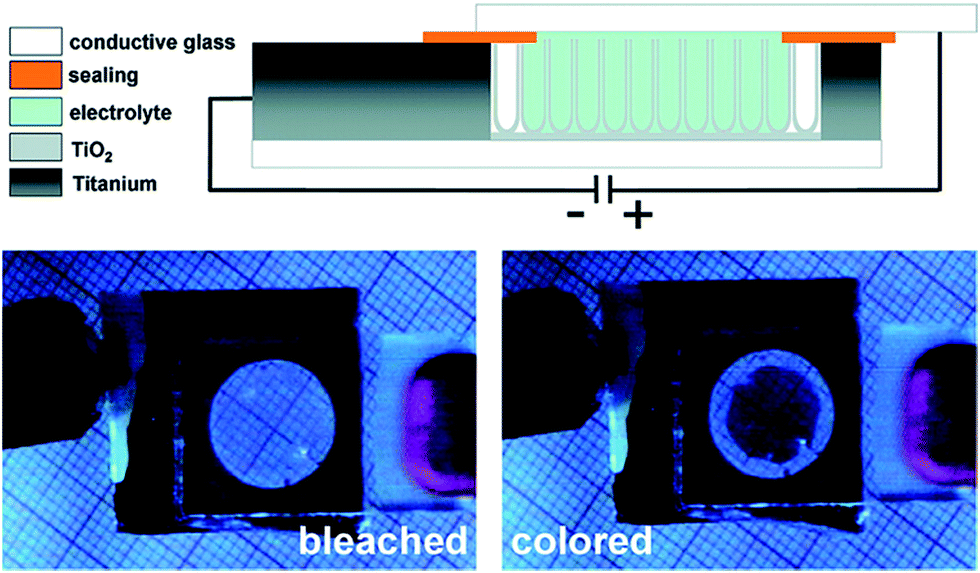 | ||
| Fig. 36 Schematic illustration and optical images of the electrochromic device based on transparent TiO2 nanotube film on conducting glass substrate in the different coloration (bleached or colored) states.255 | ||
4.5. Self-cleaning, oil–water separation and biomedical application
Since the report of TiO2 with photocatalytic abilities under UV light illumination and photo-induced superhydrophilic properties, numerous promising applications have been focused based on these materials. Organic pollutants would be photocatalytically degraded and brought away by water layer on superhydrophilic TiO2 substrates (Fig. 37a and b).257 Utilizing this strategy, a wide variety of superhydrophilic TiO2-based materials have been constructed, exhibiting environmental friendly applications for self-cleaning, anti-fogging, underwater superoleophobicity, etc. Lee et al. constructed multi-layer composite coatings with superhydrophilic, self-cleaning and anti-fogging/reflection abilities with a layer-by-layer deposition of TiO2 and SiO2 nanoparticles.258 The presence of nanoporous TiO2-based layers in composite coatings resulted in quick wicking of water into the highly hydrophilic network, exhibiting a long term superhydrophilic and anti-fogging properties (Fig. 37c–e).
 | ||
| Fig. 37 (a) Schematic depicting the self-cleaning via a photo-catalytic degradation process on a superhydrophobic TiO2 substrate covered with organic pollutants. (b) Schematic diagram of the moving of organic pollutants or dusts when water complete spreading on a superhydrophilic TiO2 substrate.257 Images of a water droplet before (c) and after (d) contacting with the superhydrophilic TiO2–SiO2 composite coating, and image demonstrating the anti-fogging abilities of TiO2-based multi-layers in composite coatings (e).258 | ||
Surface self-cleaning effect is also a significant application of superhydrophobic surface. For superhydrophobic TiO2 surfaces, the solid/water contact area is minimized. Water forms spherical droplets that easily slide off the surface and collect the dusts and photocatalytically degraded products on the way. Therefore, the superhydrophobic rough surfaces with low adhesion always exhibit a very low degree of contamination.259,260 Zhou et al. reported a long-term superhydrophobic self-cleaning coating prepared by simply blending ambient-cured fluorinated polysiloxane binder with TiO2 nanoparticles. The obtained coating exhibited excellent anti-wetting stability in various environments and photocatalytic self-cleaning property for practical applications.261 Kamegawa et al. reported superhydrophobic surfaces with photocatalytic self-cleaning properties through coating a nanocomposite TiO2 photocatalyst and hydrophobic polytetrafluoroethylene onto a structured substrate by applying a co-deposition technique.262 Recently, Parkin et al. developed a practical approach to construct robust superhydrophobic self-cleaning surfaces by taking advantage of commercial adhesive to bond the hydrophobic TiO2 nanoparticles to various substrates such as clothes, paper, glass, and steel.263 They also reported that the oil (hexadecane) contaminated surface still maintained self-cleaning ability under various environments including underwater, in air or even oil. Moreover, these treated substrates kept the excellent anti-wetting ability after finger-wipe, knife-scratch, or up to 40 sandpaper abrasion cycles.
In addition to superhydrophobicity and superhydrophilicity in air, underwater superoleophobicity is also a promising characteristic for self-cleaning surfaces, which can be easily achieved with the combination of suitable structure and high energy chemical component or constructed TiO2-based surfaces after light irradiation. In nature, fish scale and shark skin are known to be able to prevent oil contamination due to a thin water layer on their surface, exhibiting excellent underwater self-cleaning and bio-antifouling. In general, anti-fouling, low drag, slippery surfaces, and photocatalytic activities play a vital role in the underwater self-cleaning effect.264,265 Recently, Sawai et al. reported the preparation of smooth TiO2 film for sustainable underwater self-cleaning activity attributed to the photo-induced hydrophilic surface for thin water film formation to effectively prevent oils from contacting the TiO2 film.265 The research on underwater superoleophobicity is still in its infancy, and the characterization techniques for self-cleaning have to be further developed.
Besides pristine TiO2, other TiO2-based materials such as titanates have also been attracted attention for self-cleaning applications. For example, highly hydrophobic titanate nanobelt (TNB) particle can be easily prepared by a hydrogen-bond-driven assembly of pre-hydrolysed fluoroalkylsilane (FAS).266–268 Using this kind of TNB/FAS powder, a facile spinning, spraying or dipping process was applied to coat a various substrates with cross-aligned superhydrophobic film.268 The as-prepared coating displayed good self-cleaning ability and high chemical stability in a wide range of pH solutions.
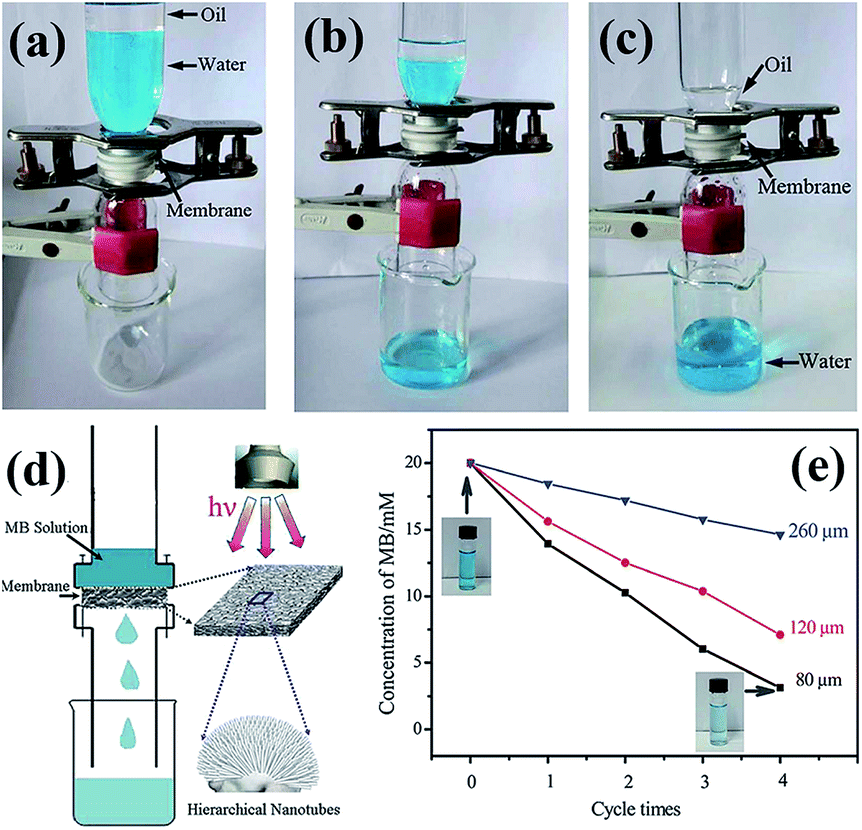 | ||
| Fig. 38 Proof-of-concept oil–water separation device and process (a–c) using anodized porous TiO2 membrane. The pre-wetted membrane was fixed between two glass vessels. A mixture of petroleum ether and water was poured into the upper vessel (a). Water permeated through the anodized porous membrane (b), while oil stayed in the upper glass vessel (c), which let to oil–water separation. (d) Schematic diagram for the flow-through photocatalysis device with anodized porous TiO2 membrane for the photocatalytic degradation of methylene blue (MB). (e) The concentration variation of MB solution after the photocatalytic degradation with various anodized porous membrane photocatalyst and circulated for different times. Inset: the photographs of MB solution before and after 4-cycles flow-through photocatalytic reaction with a maximal pore size of 80 mm.275 | ||
Recently, inspired by living creatures with special wettability and their precise arrangement of topographical structure and chemical component, we described a facile one-step approach to large scale construct superamphiphobic TiO2 particles film.278 The as-prepared TiO2 films exhibit excellent superamphiphilic property in air, changes to superamphiphobicity with good dynamical stability after silane modification (Fig. 39a). Moreover, such superamphiphobic surface reversibly can switch from superoleophobic to superoleophilic in air and underwater, respectively (Fig. 39b and c). The 3D functional surface would be a versatile platform in a wide range of applications, e.g., self-cleaning, friction reduction, and repeatable oil–water separation. In a proof-of-concept study, we applied the fluoroalkylsilane-modified pinecone-like TiO2 particles samples with both superamphiphobic in air and superoleophilic underwater as “oil capture hands” to gather oil droplets in water (Fig. 39d, process 1–3). The aggregated oils were then extracted to achieve effective oil–water separation (process 4–6). More importantly, it is worth noting that the sample maintain self-cleaning without any oil residue because of the excellent superamphiphobic property in air. This is different from the conventional superhydrophobic surface that is completely oil contaminated after oil absorption in water, which makes it difficult for repeated use. This study opens up a new avenue for the treatment of oily waste water and points out a promising application in industrial fields.
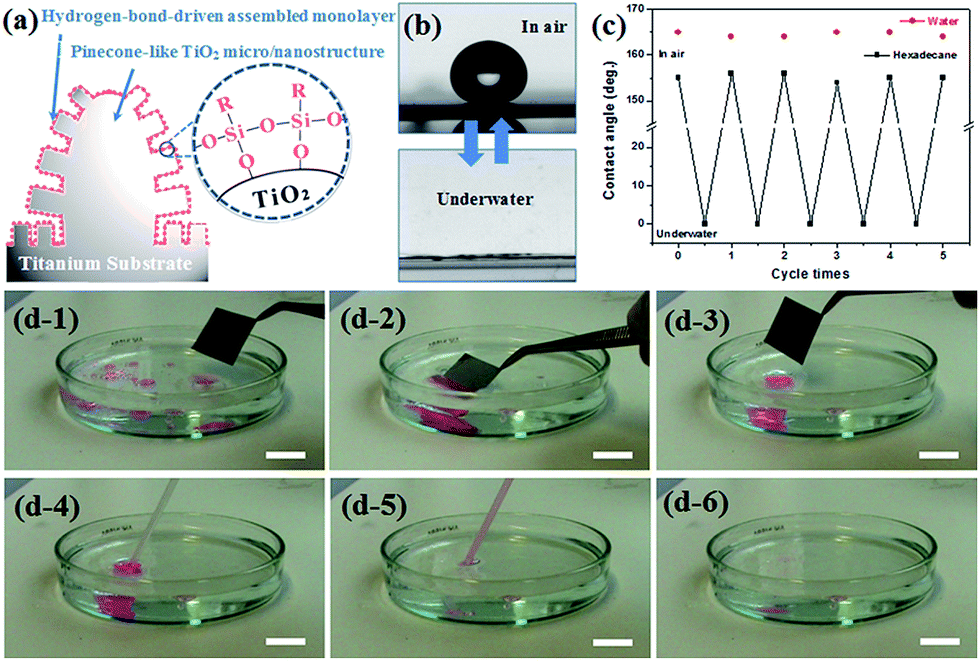 | ||
| Fig. 39 (a) The hydrogen-bonding-driven self-assembled process on the hydroxylated TiO2 particles surface. (b) The oil droplet image on the modified TiO2 surface in air or underwater environment. (c) The reversible wettability change of oil droplet on the TiO2 surface by alternating the environment. (d) The oil capture and collection process with a superoleophobic plate and underwater superoleophilic property. Process 1–3: a superoleophobic plate touches, captures and collects the sprayed oil drops in bottom of a glass container. Process 4–6: the gathered oil droplets are collected from the water. The oil was colored pink for convenient observation through the capture and collection process.278 | ||
As discussed above, multifunctional TiO2 surfaces with special wettability and adhesion have attracted great amount of attention partially owing to their promising applications in anti-bioadhesion, self-cleaning, oil–water separation, and photocatalysis. Recently, Huang et al.279 constructed a TiO2@fabric hierarchical composite via a facile strategy for loading TiO2 nanoparticles through a one-pot hydrothermal reaction (Fig. 40). By taking advantage of the excellent superhydrophobic property with UV-resistance, low droplet sliding angle and high contrast of wettability for oil and water, the as-constructed UV-durable TiO2@fabrics pave the way for their practical applications in various fields such as UV-shielding, self-cleaning and oil–water separation.
 | ||
| Fig. 40 Schematic representation of robust superhydrophobic TiO2@fabrics prepared via a facile one-pot hydrothermal strategy for hierarchical TiO2 particles construction and fluoroalkylsilane modification as a multi-functional platform for UV shielding, self-cleaning, and oil–water separation.279 | ||
Platelet adhesion and activation on biomedical material surfaces may result in blood coagulation and thrombosis. Therefore, regulating the wettability to increase the blood-compatibility is an efficient way for platelet anti-adhesion. Recently, Yang and Huang et al. combined superhydrophobicity with photocatalysis by coating a TiO2 surface with fluoroalkylsilane to induce anti-platelet effect.296,297 The in vitro blood compatibility experiment indicates that the superhydrophobic TiO2 nanotube layers exhibit a remarkable ability in reducing platelets adhesion. Compared to the abundant platelets adhered on smooth stainless steel surface (Fig. 41a and b), relatively low amount of platelets entrapped into fibrin network on hydrophilic rough TiO2 nanotube arrays upon stainless steel substrate, high TiO2 layers (Fig. 41c and d), and only very few platelets were found to adhere on the superhydrophobic TiO2 nanotube layers (Fig. 41e and f).298 Moreover, even though some platelets were occasionally attached on the superhydrophobic surface, they looked smooth without any growth of pseudopods, implying that the platelets adhered on the superhydrophobic TiO2 nanotube surface remain inactive and hardly grow or spread out. From the in vitro evaluation, the superhydrophobic TiO2 layers exhibit excellent blood compatibility and remarkable performance in preventing platelets or bacterials from adhering to the implant surface due to the creation of nanobubbles or microbubbles so that the direct attachment of cells on substrate is reduced (Fig. 41g and h). Tang et al. also observed similar bacterial adhesion behaviour on TiO2 structures with various surface wettability.299 Compared to ordinary hydrophilic or hydrophobic film, the as-prepared super-antiwetting film can greatly decrease the adhesion and activation of Staphylococcus aureus. This indicates that the special topography and wettability play critical role in blood-compatibility. Attempts for developing hybrid anti-bacterial materials by combining superhydrophobicity with anti-bacterial metals or with photocatalysis have been verified to enhance the anti-bacterial ability while maintaining the primary function of preventing bacterial adhesion. The effort of loading anti-bacterial metallic ions (e.g., Ag, Cu and Mo) with superhydrophobicity has been more successful than combining photocatalysis with superhydrophobicity. In another effort, Lai et al. demonstrated that the combined super-antiwetting and photocatalysis on TiO2 surface, together with loading of Ag particles would largely enhance the antibacterial activity.300
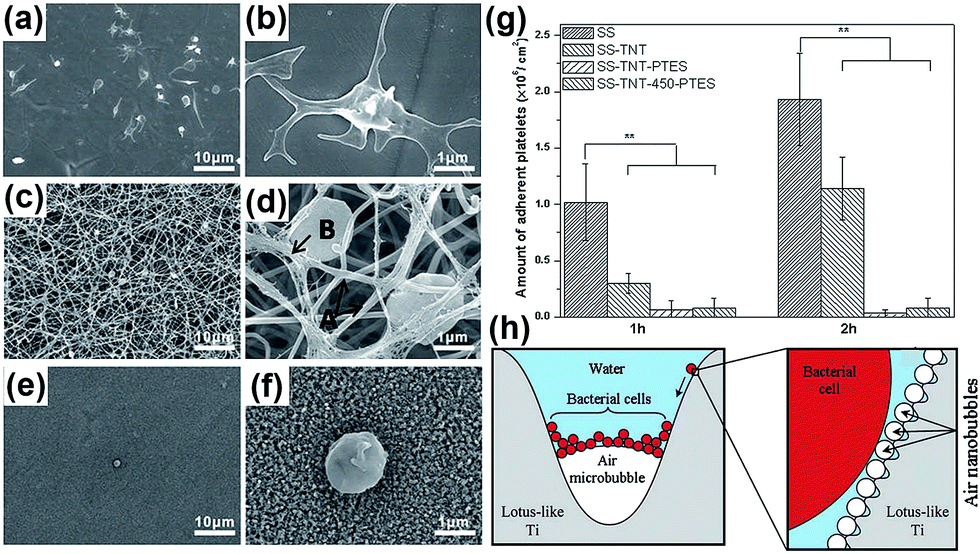 | ||
| Fig. 41 SEM images of adherent platelets on the surface of stainless steel (a and b), superhydrophilic TiO2 nanotube film on stainless steel (c and d), and superhydrophobic modified TiO2 nanotube film on stainless steel (e and f). (g) Number of adherent platelets on different samples.297 (f) Proposed mechanism by which bacterial cells accumulate at the tri-phase interface on immersed superhydrophobic Ti surfaces.298 | ||
On the basis of photocatalytic lithography patterning technique on TiO2 structures, two dimensional (2D) superhydrophilic–superhydrophobic pattern has been prepared to guide the selective deposition of bioactive calcium phosphate (CaP) layer (Fig. 42a–c), and further induce the cell attachment onto the CaP/TiO2 nanotube film (Fig. 42d–f).300–302 It can be clearly seen that CaP nanobelts are preferred to be deposited within the superhydrophilic regions to form CaP pattern while the superhydrophobic areas are kept intact. In the case of the bioadhesion, cells preferentially attached and activated on CaP/TiO2 regions to generate 3D pattern. Following this principle, arrays of TiO2, CdS, ZnO, Ag and molecular probes303–305 were assembled directly from solution onto Ti substrates at the desired locations precisely. These patterned structures can then be fabricated into arrays of photodetector, gas sensor or matrix devices for large-area microelectronic applications or bio-compatible coatings where drugs can be encapsulated in specific areas of the coating.306 This patterning strategy will be helpful to develop various micropatterned functional nanocomposite materials. However, a great challenge for wide-spread practical applications still remains to be the development of high-throughput, low-cost, and easily-controllable techniques to achieve desired orientation and order of the nano-blocks.
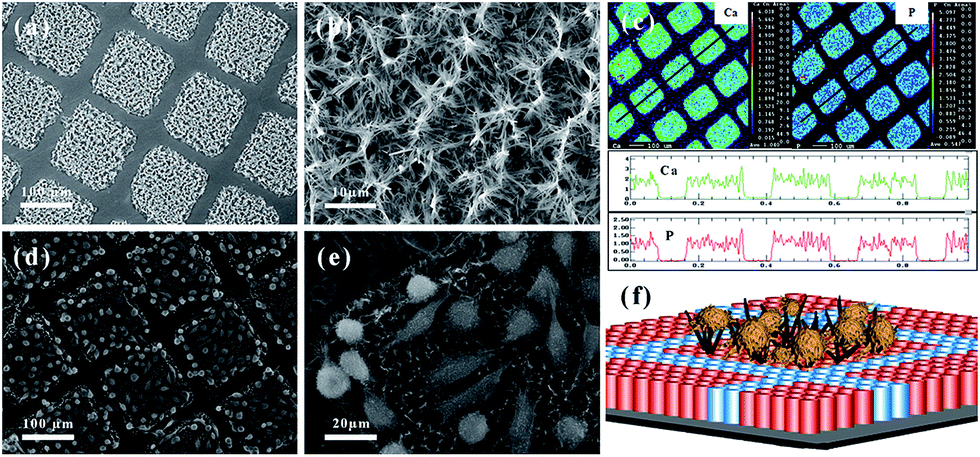 | ||
| Fig. 42 (a) Typical SEM images of the 3D spatially patterned CaP micro-nanostructure films on TiO2 film. (b) Magnified SEM of CaP crystals on superhydrophilic region. (c) Element distribution maps and corresponding line-scan signal intensity profiles of Ca and P elements across the square pattern (black line direction). (d and e) SEM images of the MG-63 osteosarcoma cells adhering on the CaP patterning surface. (f) Schematic diagram of the site-selective cell immobilization on 3D rough CaP@TiO2 scaffold.300 | ||
5. Conclusions and perspectives
In this review, we present the state-of-the-art of the development of TiO2-based materials, namely titanate and titania, for environmental and energy applications. The titanate and titania materials can be easily transformed to each other via simple processes. For example, titanate materials can be transformed to titania via thermal annealing or hydrothermal treatment of hydrogen titanate, and the titania material can react with the alkaline solutions to form titanate materials. A clear advantage of synthesizing titania from titanate is to maintain or make use of the various nanostructures easily obtainable for titanate materials. Guided by the basic formation principles of bulk materials by solid-state reaction, the preparation strategies of the formation of different dimensional titanate materials via liquid- or vapor-phase hydrothermal reaction, external and internal modified hydrothermal reaction, molten-salt reaction, high temperature oxidation, and electrochemical spark discharge spallation (ESDS) process are summarized in this review. Among them, hydrothermal route is most popular for the synthesis of 1D, 2D and 3D titanate materials due to the easy operation and rational control of the nanostructures.Due to the unique crystal structure and physical–chemical properties, titanate materials show great potential for applications in adsorption, lithium ion batteries, biomedical applications, self-cleaning, and oil–water separation. Since titanate materials possess large interlayer spacing and adjustable lattice parameters, they can be widely used as adsorbents due to their unique ion-exchange property. To be a good adsorbent, the adsorption capacity and removal rate are important considerations. The adsorption performance depends on the surface area and the number of exchangeable cations in the titanate materials.
For the photocatalytic application, due to their wide band gap (3.4–3.7 eV) and the crystal defects, the photocatalytic performance of titanate materials is not as good as TiO2. Thus, it is essential to obtain the titania materials with required crystal structure and designed nano-construction from titanate by rational control of the morphology of the starting material. An interesting strategy is to combine the excellent adsorption ability of titanates and high photocatalytic activity of titania to promote faster photodegradation of organic pollutants in wastewater. Also, the doping technology and hybrid structures are also introduced to enhance the visible light activity.
For the energy storage application, titanate and titania materials show good potential in replacing the commercial carbon negative electrodes, offering excellent rate capacities and ultralong cycle life with enhanced safety benefited from the high lithiation potential, and absence of the lithium electroplating and the charge loss problem arising from the formation of a solid–electrolyte interface layer. As an important photoanode for the DSSC and perovskite solar cells, the engineering of TiO2 nanostructures towards prolonging the charge carrier lifetime, and bandgap matching with dye or (organometal halides) are critical to achieve the goal of high efficiency and low cost in the utilization of solar energy. By making use of the reversible transition from Ti4+ to Ti3+ into TiO2 host by ion intercalation/de-intercalation, the visible electrochromism effect can be achieved on the TiO2 materials.
Lastly, multifunctional TiO2-based surfaces with special wettability and adhesion have attracted great attention owing to their promising applications in anti-bioadhesion, self-cleaning, oil–water separation.
Overall, substantial work has been carried out in the synthesis of titanate and titania nanostructures and their applications. Excellent proof-of-concept work has been realized at laboratory scale. Future effort should be focused on the improvement of its functionalities through materials and structural optimization, eventually leading to practical environmental, energy, and other related applications. Practical application of titanate and titania materials is realistically achievable as these materials are intrinsically stable in a wide range of chemical media and under UV illumination, earth-abundant, and low cost in production. For the absorbent application, the mass production on titanate materials with high surface area is an important future effort towards low-cost and effective pollutant removal, especially for the toxic metal ions and non-degradable organic dye. Little attention has been paid so far on the disposal or recovery of the titanate materials (together with some metal ions of high value) after adsorption. More research effort should be made in this aspect the future.
As a well-studied photocatalyst system in the past decades, the photocatalytic efficiency of TiO2 under full solar energy spectrum is still low. New photoconversion concept based on rational materials design or bandgap engineering is urgently needed to achieve high solar-to-fuel conversion efficiency. For other energy related applications, much effort is needed to commercialize these TiO2-based materials for rechargeable lithium ion battery and DSSC applications. In particular, the exploration on the TiO2 materials towards solid state, wearable and printable energy devices will have a promising future for the development of efficient and flexible optoelectronics. For the wetting ability control on TiO2 nanostructure, the rate of wetting property change, stability under UV, and mechanical strength should be carefully studied in the future.
Acknowledgements
The authors thank the Environment and Water Industry Programme Office (EWI) under the National Research Foundation (NRF) of Singapore (grant MEWR651/06/160), National Natural Science Foundation of China (grant 21501127; 51502185), Natural Science Foundation of Jiangsu Province of China (grant BK20130313), Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) for financial support of this work.References
- M. L. McKinney, R. M. Schoch and L. Yonavjak, Environmental Science: Systems and Solutions, Jones and Barlett Publishers, 2012 Search PubMed
.
- E. G. Snyder, T. H. Watkins, P. A. Solomon, E. D. Thoma, R. W. Williams, G. S. W. Hagler, D. Shelow, D. A. Hindin, V. J. Kilaru and P. W. Preuss, Environ. Sci. Technol., 2013, 47, 11369–11377 CrossRef CAS PubMed
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148–173 CAS
- P. Xu, Y. Chen and X. Ye, Lancet, 2013, 382, 2067 CrossRef
- X. Lang, X. Chen and J. Zhao, Chem. Soc. Rev., 2014, 43, 473–486 RSC
- D. Fattakhova-Rohlfing, A. Zaleska and T. Bein, Chem. Rev., 2014, 114, 9487–9558 CrossRef CAS PubMed
- X. Wang, Z. Li, J. Shi and Y. Yu, Chem. Rev., 2014, 114, 9346–9384 CrossRef CAS PubMed
- A. K. Chandiran, A. Yella, M. T. Mayer, P. Gao, M. K. Nazeeruddin and M. Graetzel, Adv. Mater., 2014, 26, 4309–4312 CrossRef CAS PubMed
- J. Kim, H. Lee, D. Y. Kim and Y. Seo, Adv. Mater., 2014, 26, 5192–5197 CrossRef CAS PubMed
- Y. Tang, Y. Zhang, J. Deng, J. Wei, H. L. Tam, B. K. Chandran, Z. Dong, Z. Chen and X. Chen, Adv. Mater., 2014, 26, 6111–6118 CrossRef CAS PubMed
- J. Fei and J. Li, Adv. Mater., 2015, 27, 314–319 CrossRef CAS PubMed
- H. Liu, Z. Bi, X.-G. Sun, R. R. Unocic, M. P. Paranthaman, S. Dai and G. M. Brown, Adv. Mater., 2011, 23, 3450–3454 CrossRef CAS PubMed
- S. Xiong, Y. Tang, H. S. Ng, X. Zhao, Z. Jiang, Z. Chen, K. W. Ng and S. C. J. Loo, Toxicology, 2013, 304, 132–140 CrossRef CAS PubMed
- D. V. Bavykin, J. M. Friedrich and F. C. Walsh, Adv. Mater., 2006, 18, 2807–2824 CrossRef CAS PubMed
- N. Liu, X. Chen, J. Zhang and J. W. Schwank, Catal. Today, 2014, 225, 34–51 CrossRef CAS PubMed
- Y. L. Pang, S. Lim, H. C. Ong and W. T. Chong, Appl. Catal., A, 2014, 481, 127–142 CrossRef CAS PubMed
- H. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS
- Q. Tay, X. Liu, Y. Tang, Z. Jiang, T. C. Sum and Z. Chen, J. Phys. Chem. C, 2013, 117, 14973–14982 CAS
- D. V. Bavykin, M. Carravetta, A. N. Kulak and F. C. Walsh, Chem. Mater., 2010, 22, 2458–2465 CrossRef CAS
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Adv. Mater., 1999, 11, 1307–1311 CrossRef CAS
- G. H. Du, Q. Chen, R. C. Che, Z. Y. Yuan and L.-M. Peng, Appl. Phys.
Lett., 2001, 79, 3702–3704 CrossRef CAS PubMed
- Q. Chen, G. H. Du, S. Zhang and L.-M. Peng, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 587–593 CrossRef CAS PubMed
- R. Ma, Y. Bando and T. Sasaki, Chem. Phys. Lett., 2003, 380, 577–582 CrossRef CAS PubMed
- T. Gao, H. Fjellvåg and P. Norby, Chem. Mater., 2009, 21, 3503–3513 CrossRef CAS
- N. Masaki, S. Uchida and T. Sato, J. Mater. Chem., 2002, 12, 305–308 RSC
- H. Izawa, S. Kikkawa and M. Koizumi, J. Phys. Chem., 1982, 86, 5023–5026 CrossRef CAS
- T. Sasaki, K. M. Yu and Y. Fujiki, Chem. Mater., 1992, 4, 894–899 CrossRef CAS
- R. Marchand, L. Brohan and M. Tournoux, Mater. Res. Bull., 1980, 15, 1129–1133 CrossRef CAS
- J. S. Wang, S. Yin and T. Sato, Chem. Lett., 2004, 33, 1104–1105 CrossRef CAS
- Y. H. Cheng, Y. Huang, P. D. Kanhere, V. P. Subramaniam, D. Gong, S. Zhang, J. Highfield, M. K. Schreyer and Z. Chen, Chem.–Eur. J., 2011, 17, 2575–2578 CrossRef CAS PubMed
- Y. H. Cheng, D. Gong, Y. Tang, J. W. C. Ho, Y. Y. Tay, W. S. Lau, O. Wijaya, J. Lim and Z. Chen, J. Solid State Chem., 2014, 214, 67–73 CrossRef CAS PubMed
- Y. Ren, Z. Liu, F. Pourpoint, A. R. Armstrong, C. P. Grey and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 2164–2167 CrossRef CAS PubMed
- Q. Chen, W. Z. Zhou, G. H. Du and L. M. Peng, Adv. Mater., 2002, 14, 1208–1211 CrossRef CAS
- T. Gao, H. Fjellvag and P. Norby, Inorg. Chem., 2009, 48, 1423–1432 CrossRef CAS PubMed
- V. B. Dmitry, et al., Nanotechnology, 2008, 19, 275604 CrossRef PubMed
- D. V. Bavykin and F. C. Walsh, Eur. J. Inorg. Chem., 2009, 977–997 CrossRef CAS PubMed
- D. V. Bavykin, V. N. Parmon, A. A. Lapkin and F. C. Walsh, J. Mater. Chem., 2004, 14, 3370–3377 RSC
- Y. Lan, X. P. Gao, H. Y. Zhu, Z. F. Zheng, T. Y. Yan, F. Wu, S. P. Ringer and D. Y. Song, Adv. Funct. Mater., 2005, 15, 1310–1318 CrossRef CAS PubMed
- X. Sun, X. Chen and Y. Li, Inorg. Chem., 2002, 41, 4996–4998 CrossRef CAS PubMed
- W. A. Daoud and G. K. H. Pang, J. Phys. Chem. B, 2006, 110, 25746–25750 CrossRef CAS PubMed
- Y. W. L. Lim, Y. Tang, Y. H. Cheng and Z. Chen, Nanoscale, 2010, 2, 2751–2757 RSC
- D. L. Morgan, H. Y. Zhu, R. L. Frost and E. R. Waclawik, Chem. Mater., 2008, 20, 3800–3802 CrossRef CAS
- S. Zhang, L. M. Peng, Q. Chen, G. H. Du, G. Dawson and W. Z. Zhou, Phys. Rev. Lett., 2003, 91, 256103 CrossRef CAS
- N. Viriya-empikul, T. Charinpanitkul, N. Sano, A. Soottitantawat, T. Kikuchi, K. Faungnawakij and W. Tanthapanichakoon, Mater. Chem. Phys., 2009, 118, 254–258 CrossRef CAS PubMed
- Y. Zhu, H. Li, Y. Koltypin, Y. R. Hacohen and A. Gedanken, Chem. Commun., 2001, 2616–2617 RSC
- Y. Ma, Y. Lin, X. Xiao, X. Zhou and X. Li, Mater. Res. Bull., 2006, 41, 237–243 CrossRef CAS PubMed
- H.-H. Ou, C.-H. Liao, Y.-H. Liou, J.-H. Hong and S.-L. Lo, Environ. Sci. Technol., 2008, 42, 4507–4512 CrossRef CAS
- F. Dufour, S. Cassaignon, O. Durupthy, C. Colbeau-Justin and C. Chanéac, Eur. J. Inorg. Chem., 2012, 2012, 2707–2715 CrossRef CAS PubMed
- L. Zhu, L. Gu, Y. Zhou, S. Cao and X. Cao, J. Mater. Chem., 2011, 21, 12503–12510 RSC
- X. Cao, Y. Zhou, J. Wu, Y. Tang, L. Zhu and L. Gu, Nanoscale, 2013, 5, 3486–3495 RSC
- E. Horvath, A. Kukovecz, Z. Konya and I. Kiricsi, Chem. Mater., 2007, 19, 927–931 CrossRef CAS
- L. Torrente-Murciano, A. A. Lapkin and D. Chadwick, J. Mater. Chem., 2010, 20, 6484–6489 RSC
- Y. Tang, Y. Zhang, J. Deng, D. Qi, W. R. Leow, J. Wei, S. Yin, Z. Dong, R. Yazami, Z. Chen and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 13488–13492 CrossRef CAS PubMed
- C. Y. Xu, Q. Zhang, H. Zhang, L. Zhen, J. Tang and L. C. Qin, J. Am. Chem. Soc., 2005, 127, 11584–11585 CrossRef CAS PubMed
- Z. Liu, T. Yamazaki, Y. Shen, D. Meng, T. Kikuta and N. Nakatani, J. Phys. Chem. C, 2008, 112, 4545–4549 CAS
- D. P. Macwan, P. Dave and S. Chaturvedi, J. Mater. Sci., 2011, 46, 3669–3686 CrossRef CAS
- D. Li and Y. Xia, Nano Lett., 2003, 3, 555–560 CrossRef CAS
- J. T. McCann, D. Li and Y. Xia, J. Mater. Chem., 2005, 15, 735–738 RSC
- D. Li and Y. Xia, Nano Lett., 2004, 4, 933–938 CrossRef CAS
- J. Wang, J. Hou, M. W. Ellis and A. S. Nain, New J. Chem., 2013, 37, 571–574 RSC
- C. K. Kwok and S. B. Desu, Appl. Phys. Lett., 1992, 60, 1430–1432 CrossRef CAS PubMed
- Z. Chang, J. Liu, J. F. Liu and X. M. Sun, J. Mater. Chem., 2011, 21, 277–282 RSC
- B. Zhao, F. Chen, X. N. Gu and J. L. Zhang, Chem.–Asian J., 2010, 5, 1546–1549 CrossRef CAS PubMed
- Z. Zhang, J. B. M. Goodall, S. Brown, L. Karlsson, R. J. H. Clark, J. L. Hutchison, I. U. Rehman and J. A. Darr, Dalton Trans., 2010, 39, 711–714 RSC
- N. Sutradhar, A. Sinhamahapatra, S. Kumar Pahari, H. C. Bajaj and A. Baran Panda, Chem. Commun., 2011, 47, 7731–7733 RSC
- N. Kijima, M. Kuwabara, J. Akimoto, T. Kumagai, K. Igarashi and T. Shimizu, J. Power Sources, 2011, 196, 7006–7010 CrossRef CAS PubMed
- T. Sasaki and M. Watanabe, J. Am. Chem. Soc., 1998, 120, 4682–4689 CrossRef CAS
- J. L. Gunjakar, T. W. Kim, H. N. Kim, I. Y. Kim and S.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 14998–15007 CrossRef CAS PubMed
- R. Ma, T. Sasaki and Y. Bando, J. Am. Chem. Soc., 2004, 126, 10382–10388 CrossRef CAS PubMed
- N. Sakai, K. Fukuda, Y. Omomo, Y. Ebina, K. Takada and T. Sasaki, J. Phys. Chem. C, 2008, 112, 5197–5202 CAS
- H. N. Kim, T. W. Kim, I. Y. Kim and S.-J. Hwang, Adv. Funct. Mater., 2011, 21, 3111–3118 CrossRef CAS PubMed
- T. W. Kim, S. G. Hur, S. J. Hwang, H. Park, W. Choi and J. H. Choy, Adv. Funct. Mater., 2007, 17, 307–314 CrossRef CAS PubMed
- J. Q. Huang, Z. Huang, W. Guo, M. L. Wang, Y. G. Cao and M. C. Hong, Cryst. Growth Des., 2008, 8, 2444–2446 CAS
- J. Jitputti, T. Rattanavoravipa, S. Chuangchote, S. Pavasupree, Y. Suzuki and S. Yoshikawa, Catal. Commun., 2009, 10, 378–382 CrossRef CAS PubMed
- Y. Takezawa and H. Imai, Small, 2006, 2, 390–393 CrossRef CAS PubMed
- Y. B. Mao, M. Kanungo, T. Hemraj-Benny and S. S. Wong, J. Phys. Chem. B, 2006, 110, 702–710 CrossRef CAS PubMed
- C. W. Peng, T. Y. Ke, L. Brohan, M. Richard-Plouet, J. C. Huang, E. Puzenat, H. T. Chiu and C. Y. Lee, Chem. Mater., 2008, 20, 2426–2428 CrossRef CAS
- C. Wu, L. Lei, X. Zhu, J. Yang and Y. Xie, Small, 2007, 3, 1518–1522 CrossRef CAS PubMed
- W. Li, Y. Deng, Z. Wu, X. Qian, J. Yang, Y. Wang, D. Gu, F. Zhang, B. Tu and D. Zhao, J. Am. Chem. Soc., 2011, 133, 15830–15833 CrossRef CAS PubMed
- Y. X. Tang, Y. K. Lai, D. G. Gong, K. H. Goh, T. T. Lim, Z. L. Dong and Z. Chen, Chem.–Eur. J., 2010, 16, 7704–7708 CrossRef CAS PubMed
- Y. X. Tang, D. G. Gong, Y. K. Lai, Y. Q. Shen, Y. Y. Zhang, Y. Z. Huang, J. Tao, C. J. Lin, Z. L. Dong and Z. Chen, J. Mater. Chem., 2010, 20, 10169–10178 RSC
- S. Uchida, Y. Yamamoto, Y. Fujishiro, A. Watanabe, O. Ito and T. Sato, J. Chem. Soc., Faraday Trans., 1997, 93, 3229–3234 RSC
- J. H. Choy, H. C. Lee, H. Jung and S. J. Hwang, J. Mater. Chem., 2001, 11, 2232–2234 RSC
- D. J. D. Corcoran, D. P. Tunstall and J. T. S. Irvine, Solid State Ionics, 2000, 136–137, 297–303 CrossRef CAS
- H. Y. Niu, J. M. Wang, Y. L. Shi, Y. Q. Cai and F. S. Wei, Microporous Mesoporous Mater., 2009, 122, 28–35 CrossRef CAS PubMed
- M. D. Wei, K. W. Wei, M. Ichihara and H. S. Zhou, Electrochem. Commun., 2008, 10, 1164–1167 CrossRef CAS PubMed
- A. D. Weisz, L. García Rodenas, P. J. Morando, A. E. Regazzoni and M. A. Blesa, Catal. Today, 2002, 76, 103–112 CrossRef CAS
- W. Liu, A. G. L. Borthwick, X. Li and J. Ni, Microporous Mesoporous Mater., 2014, 186, 168–175 CrossRef CAS PubMed
- Q. Y. Li, T. Kako and J. H. Ye, Appl. Catal., A, 2010, 375, 85–91 CrossRef CAS PubMed
- R. Z. Ma, T. Sasaki and Y. Bando, Chem. Commun., 2005, 948–950 RSC
- N. Sakai, Y. Ebina, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2004, 126, 5851–5858 CrossRef CAS PubMed
- D. V. Bavykin, A. A. Lapkin, P. K. Plucinski, J. M. Friedrich and F. C. Walsh, J. Catal., 2005, 235, 10–17 CrossRef CAS PubMed
- J. J. Yang, Z. S. Jin, X. D. Wang, W. Li, J. W. Zhang, S. L. Zhang, X. Y. Guo and Z. J. Zhang, Dalton Trans., 2003, 3898–3901 RSC
- D. J. Yang, Z. F. Zheng, H. W. Liu, H. Y. Zhu, X. B. Ke, Y. Xu, D. Wu and Y. Sun, J. Phys. Chem. C, 2008, 112, 16275–16280 CAS
- D. J. Yang, Z. F. Zheng, H. Y. Zhu, H. W. Liu and X. P. Gao, Adv. Mater., 2008, 20, 2777–2781 CrossRef CAS PubMed
- C. K. Lee, K. S. Lin, C. F. Wu, M. D. Lyu and C. C. Lo, J. Hazard. Mater., 2008, 150, 494–503 CrossRef CAS PubMed
- D. Wu, Y. Chen, J. Liu, X. Zhao, A. Li and N. Ming, Appl. Phys. Lett., 2005, 87, 112501–112503 CrossRef PubMed
- X. G. Xu, X. Ding, Q. Chen and L. M. Peng, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165403 CrossRef
- C. Huang, X. Liu, L. Kong, W. Lan, Q. Su and Y. Wang, Appl. Phys. A: Mater. Sci. Process., 2007, 87, 781–786 CrossRef CAS
- P. Z. Araujo, C. B. Mendive, L. A. G. Rodenas, P. J. Morando, A. E. Regazzoni, M. A. Blesa and D. Bahnemann, Colloids Surf., A, 2005, 265, 73–80 CrossRef CAS PubMed
- J. M. Pettibone, D. M. Cwiertny, M. Scherer and V. H. Grassian, Langmuir, 2008, 24, 6659–6667 CrossRef CAS PubMed
- J. Liu, M. Luo, Z. Yuan and A. Ping, J. Radioanal. Nucl. Chem., 2013, 298, 1427–1434 CrossRef CAS
- W. Liu, J. Ni and X. Yin, Water Res., 2014, 53, 12–25 CrossRef CAS PubMed
- A. Turki, C. Guillard, F. Dappozze, G. Berhault, Z. Ksibi and H. Kochkar, J. Photochem. Photobiol., A, 2014, 279, 8–16 CrossRef CAS PubMed
- L. Xiong, Y. Yang, J. Mai, W. Sun, C. Zhang, D. Wei, Q. Chen and J. Ni, Chem. Eng. J., 2010, 156, 313–320 CrossRef CAS PubMed
- M. Feng, W. You, Z. Wu, Q. Chen and H. Zhan, ACS Appl. Mater. Interfaces, 2013, 5, 12654–12662 CAS
- Y. Tang, Z. Jiang, Q. Tay, J. Deng, Y. Lai, D. Gong, Z. Dong and Z. Chen, RSC Adv., 2012, 2, 9406–9414 RSC
- S. Xie, B. Zheng, Q. Kuang, X. Wang, Z. Xie and L. Zheng, CrystEngComm, 2012, 14, 7715–7720 RSC
- J. Ma, F. Yu, L. Zhou, L. Jin, M. Yang, J. Luan, Y. Tang, H. Fan, Z. Yuan and J. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 5749–5760 CAS
- C.-T. Hsieh, W.-S. Fan, W.-Y. Chen and J.-Y. Lin, Sep. Purif. Technol., 2009, 67, 312–318 CrossRef CAS PubMed
- D. Bahnemann, Sol. Energy, 2004, 77, 445–459 CrossRef CAS PubMed
- A. Nakahira, W. Kato, M. Tamai, T. Isshiki, K. Nishio and H. Aritani, J. Mater. Sci., 2004, 39, 4239–4245 CrossRef CAS
- M. A. Khan, H.-T. Jung and O. B. Yang, J. Phys. Chem. B, 2006, 110, 6626–6630 CrossRef CAS PubMed
- H. Y. Zhu, X. P. Gao, Y. Lan, D. Y. Song, Y. X. Xi and J. C. Zhao, J. Am. Chem. Soc., 2004, 126, 8380–8381 CrossRef CAS PubMed
- P. Kanhere, Y. Tang, J. Zheng and Z. Chen, J. Phys. Chem. Solids, 2013, 74, 1708–1713 CrossRef CAS PubMed
- P. Kanhere, J. Nisar, Y. Tang, B. Pathak, R. Ahuja, J. Zheng and Z. Chen, J. Phys. Chem. C, 2012, 116, 22767–22773 CAS
- G.-S. Guo, C.-N. He, Z.-H. Wang, F.-B. Gu and D.-M. Han, Talanta, 2007, 72, 1687–1692 CrossRef CAS PubMed
- V. Stengl, S. Bakardjieva, J. Subrt, E. Vecernikova, L. Szatmary, M. Klementova and V. Balek, Appl. Catal., B, 2006, 63, 20–30 CrossRef CAS PubMed
- A. Fujishima, K. Hashimoto and T. Watanabe TiO2 Photocatalysis: Fundamentals and Applications, BKC, USA, 1999 Search PubMed
- C.-K. Lee, C.-C. Wang, M.-D. Lyu, L.-C. Juang, S.-S. Liu and S.-H. Hung, J. Colloid Interface Sci., 2007, 316, 562–569 CrossRef CAS PubMed
- M. Qamar, C. R. Yoon, H. J. Oh, N. H. Lee, K. Park, D. H. Kim, K. S. Lee, W. J. Lee and S. J. Kim, Catal. Today, 2008, 131, 3–14 CrossRef CAS PubMed
- A. Riss, T. Berger, H. Grothe, J. Bernardi, O. Diwald and E. Knozinger, Nano Lett., 2007, 7, 433–438 CrossRef CAS PubMed
- A. Riss, M. J. Elser, J. Bernardi and O. Diwald, J. Am. Chem. Soc., 2009, 131, 6198–6206 CrossRef CAS PubMed
- M. Hodos, E. Horvath, H. Haspel, A. Kukovecz, Z. Kanya and I. Kiricsi, Chem. Phys. Lett., 2004, 399, 512–515 CrossRef CAS PubMed
- M. Zhang, Z. Jin, J. Zhang, X. Guo, J. Yang, W. Li, X. Wang and Z. Zhang, J. Mol. Catal. A: Chem., 2004, 217, 203–210 CrossRef CAS PubMed
- J. Yu, H. Yu, B. Cheng and C. Trapalis, J. Mol. Catal. A: Chem., 2006, 249, 135–142 CrossRef CAS PubMed
- B. M. Wen, C. Y. Liu, Y. Liu and Z. Y. Zhang, J. Nanosci. Nanotechnol., 2004, 4, 1062–1066 CrossRef CAS PubMed
- D. J. Yang, H. W. Liu, Z. F. Zheng, Y. Yuan, J. C. Zhao, E. R. Waclawik, X. B. Ke and H. Y. Zhu, J. Am. Chem. Soc., 2009, 131, 17885–17893 CrossRef CAS PubMed
- H. G. Yang and H. C. Zeng, J. Am. Chem. Soc., 2005, 127, 270–278 CrossRef CAS PubMed
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed
- J. F. Banfield, D. R. Veblen and D. J. Smith, Am. Mineral., 1991, 76, 343–353 CAS
- G. Xiang, T. Li, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 6801–6803 RSC
- B. Ohtani, J.-I. Handa, S.-I. Nishimoto and T. Kagiya, Chem. Phys. Lett., 1985, 120, 292–294 CrossRef CAS
- T. A. Kandiel, A. Feldhoff, L. Robben, R. Dillert and D. W. Bahnemann, Chem. Mater., 2010, 22, 2050–2060 CrossRef CAS
- H. Z. Zhang and J. F. Banfield, J. Phys. Chem. B, 2000, 104, 3481–3487 CrossRef CAS
- M. R. Ranade, A. Navrotsky, H. Z. Zhang, J. F. Banfield, S. H. Elder, A. Zaban, P. H. Borse, S. K. Kulkarni, G. S. Doran and H. J. Whitfield, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6476–6481 CrossRef CAS PubMed
- Y. X. Tang, P. X. Wee, Y. K. Lai, X. P. Wang, D. G. Gong, P. D. Kanhere, T. T. Lim, Z. L. Dong and Z. Chen, J. Phys. Chem. C, 2012, 116, 2772–2780 CAS
- Y. X. Tang, J. Tao, H. J. Tao, T. Wu, L. Wang, Y. Y. Zhang, Z. L. Li and X. L. Tian, Acta Phys.-Chim. Sin., 2008, 24, 1120–1126 CAS
- H. G. Yang, C. H. Sun, S. Z. Qiao, J. Zou, G. Liu, S. C. Smith, H. M. Cheng and G. Q. Lu, Nature, 2008, 453, 638–641 CrossRef CAS PubMed
- Y. Alivov and Z. Y. Fan, J. Phys. Chem. C, 2009, 113, 12954–12957 CAS
- D. H. Chen, F. Z. Huang, Y. B. Cheng and R. A. Caruso, Adv. Mater., 2009, 21, 2206–2210 CrossRef CAS PubMed
- Y. Q. Dai, C. M. Cobley, J. Zeng, Y. M. Sun and Y. N. Xia, Nano Lett., 2009, 9, 2455–2459 CrossRef CAS PubMed
- X. G. Han, Q. Kuang, M. S. Jin, Z. X. Xie and L. S. Zheng, J. Am. Chem. Soc., 2009, 131, 3152–3153 CrossRef CAS PubMed
- G. Liu, H. G. Yang, X. W. Wang, L. N. Cheng, H. F. Lu, L. Z. Wang, G. Q. Lu and H. M. Cheng, J. Phys. Chem. C, 2009, 113, 21784–21788 CAS
- G. Liu, H. G. Yang, X. W. Wang, L. N. Cheng, J. Pan, G. Q. Lu and H. M. Cheng, J. Am. Chem. Soc., 2009, 131, 12868–12869 CrossRef CAS PubMed
- H. G. Yang, G. Liu, S. Z. Qiao, C. H. Sun, Y. G. Jin, S. C. Smith, J. Zou, H. M. Cheng and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 4078–4083 CrossRef CAS PubMed
- Z. K. Zheng, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai, M. H. Jiang, P. Wang and M. H. Whangbo, Chem.–Eur. J., 2009, 15, 12576–12579 CrossRef CAS PubMed
- F. Dufour, S. Pigeot-Remy, O. Durupthy, S. Cassaignon, V. Ruaux, S. Torelli, L. Mariey, F. Maugé and C. Chanéac, Appl. Catal., B, 2015, 174–175, 350–360 CrossRef CAS PubMed
- D. Wang, P. Kanhere, M. Li, Q. Tay, Y. Tang, Y. Huang, T. C. Sum, N. Mathews, T. Sritharan and Z. Chen, J. Phys. Chem. C, 2013, 117, 22894–22902 CAS
- M. D'Arienzo, J. Carbajo, A. Bahamonde, M. Crippa, S. Polizzi, R. Scotti, L. Wahba and F. Morazzoni, J. Am. Chem. Soc., 2011, 133, 17652–17661 CrossRef PubMed
- M. D'Arienzo, M. V. Dozzi, M. Redaelli, B. D. Credico, F. Morazzoni, R. Scotti and S. Polizzi, J. Phys. Chem. C, 2015, 119, 12385–12393 Search PubMed
- T. Ohno, K. Sarukawa, K. Tokieda and M. Matsumura, J. Catal., 2001, 203, 82–86 CrossRef CAS
- T. Ohno, K. Tokieda, S. Higashida and M. Matsumura, Appl. Catal., A, 2003, 244, 383–391 CrossRef CAS
- M. A. Henderson, Surf. Sci. Rep., 2011, 66, 185–297 CrossRef CAS PubMed
- Q. A. Xu, Y. Ma, J. Zhang, X. L. Wang, Z. C. Feng and C. Li, J. Catal., 2011, 278, 329–335 CrossRef CAS PubMed
- Z. F. Zheng, H. W. Liu, J. P. Ye, J. C. Zhao, E. R. Waclawik and H. Y. Zhu, J. Mol. Catal. A: Chem., 2010, 316, 75–82 CrossRef CAS PubMed
- Z. Jiang, Y. Tang, Q. Tay, Y. Zhang, O. I. Malyi, D. Wang, J. Deng, Y. Lai, H. Zhou, X. Chen, Z. Dong and Z. Chen, Adv. Energy Mater., 2013, 3, 1368–1380 CrossRef CAS PubMed
- C. Liu, T. Sun, L. Wu, J. Liang, Q. Huang, J. Chen and W. Hou, Appl. Catal., B, 2015, 170, 17–24 CrossRef PubMed
- X. P. Wang, Y. X. Tang, M. Y. Leiw and T. T. Lim, Appl. Catal., A, 2011, 409, 257–266 CrossRef PubMed
- G. Liu, L. Z. Wang, C. H. Sun, X. X. Yan, X. W. Wang, Z. G. Chen, S. C. Smith, H. M. Cheng and G. Q. Lu, Chem. Mater., 2009, 21, 1266–1274 CrossRef CAS
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, J. Phys. Chem. Solids, 2002, 63, 1909–1920 CrossRef CAS
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed
- V. Gombac, L. de Rogatis, A. Gasparotto, G. Vicario, T. Montini, D. Barreca, G. Balducci, P. Fornasiero, E. Tondello and M. Graziani, Chem. Phys., 2007, 339, 111–123 CrossRef CAS PubMed
- N. Lu, H. M. Zhao, J. Y. Li, X. Quan and S. Chen, Sep. Purif. Technol., 2008, 62, 668–673 CrossRef CAS PubMed
- F. Dong, W. R. Zhao and Z. B. Wu, Nanotechnology, 2008, 19, 10 Search PubMed
- X. B. Chen and C. Burda, J. Am. Chem. Soc., 2008, 130, 5018–5019 CrossRef CAS PubMed
- Y. K. Lai, J. Y. Huang, H. F. Zhang, V. P. Subramaniam, Y. X. Tang, D. G. Gong, L. Sundar, L. Sun, Z. Chen and C. J. Lin, J. Hazard. Mater., 2010, 184, 855–863 CrossRef CAS PubMed
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed
- D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed., 2008, 47, 9975–9978 CrossRef CAS PubMed
- D. Mitoraj and H. Kisch, Chem.–Eur. J., 2010, 16, 261–269 CrossRef CAS PubMed
- D. Mitoraj and H. Kisch, Solid State Phenom., 2010, 162, 49–75 CrossRef CAS
- D. Gong, J. G. Highfield, S. Z. E. Ng, Y. Tang, W. C. J. Ho, Q. Tay and Z. Chen, ACS Sustainable Chem. Eng., 2014, 2, 149–157 CrossRef CAS
- Y. H. Cheng, V. P. Subramaniam, D. G. Gong, Y. X. Tang, J. Highfield, S. O. Pehkonen, P. Pichat, M. K. Schreyer and Z. Chen, J. Solid State Chem., 2012, 196, 518–527 CrossRef CAS PubMed
- H. Kisch, Angew. Chem., Int. Ed., 2013, 52, 812–847 CrossRef CAS PubMed
- Y. Zhang, Y. Tang, X. Liu, Z. Dong, H. H. Hng, Z. Chen, T. C. Sum and X. Chen, Small, 2013, 9, 996–1002 CrossRef CAS PubMed
- P. Hoyer and R. Konenkamp, Appl. Phys. Lett., 1995, 66, 349–351 CrossRef CAS PubMed
- D. Fitzmaurice, H. Frei and J. Rabani, J. Phys. Chem., 1995, 99, 9176–9181 CrossRef CAS
- R. Vogel, P. Hoyer and H. Weller, J. Phys. Chem., 1994, 98, 3183–3188 CrossRef CAS
- A. L. Linsebigler, G. Q. Lu and J. T. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS
- A. Sclafani, M. N. Mozzanega and P. Pichat, J. Photochem. Photobiol., A, 1991, 59, 181–189 CrossRef CAS
- Y. Tian and T. Tatsuma, Chem. Commun., 2004, 1810–1811 RSC
- Y. Tian and T. Tatsuma, J. Am. Chem. Soc., 2005, 127, 7632–7637 CrossRef CAS PubMed
- A. W. Zhu, Y. P. Luo and Y. Tian, Anal. Chem., 2009, 81, 7243–7247 CrossRef CAS PubMed
- D. Gong, W. C. J. Ho, Y. Tang, Q. Tay, Y. Lai, J. G. Highfield and Z. Chen, J. Solid State Chem., 2012, 189, 117–122 CrossRef CAS PubMed
- C. Clavero, Nat. Photonics, 2014, 8, 95–103 CrossRef CAS PubMed
- Y. Tian, H. Notsu and T. Tatsuma, Photochem. Photobiol. Sci., 2005, 4, 598–601 CAS
- S. K. Cushing, J. T. Li, F. K. Meng, T. R. Senty, S. Suri, M. J. Zhi, M. Li, A. D. Bristow and N. Q. Wu, J. Am. Chem. Soc., 2012, 134, 15033–15041 CrossRef CAS PubMed
- X. Chen, H. Y. Zhu, J. C. Zhao, Z. T. Zheng and X. P. Gao, Angew. Chem., Int. Ed., 2008, 47, 5353–5356 CrossRef CAS PubMed
- X. Chen, Z. F. Zheng, X. B. Ke, E. Jaatinen, T. F. Xie, D. J. Wang, C. Guo, J. C. Zhao and H. Y. Zhu, Green Chem., 2010, 12, 414–419 RSC
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC
- Y. K. Lai, H. F. Zhuang, K. P. Xie, D. G. Gong, Y. X. Tang, L. Sun, C. J. Lin and Z. Chen, New J. Chem., 2010, 34, 1335–1340 RSC
- K. F. Yu, Y. Tian and T. Tatsuma, Phys. Chem. Chem. Phys., 2006, 8, 5417–5420 RSC
- Y. Tian, X. T. Wang, D. Zhang, X. Shi and S. L. Wang, J. Photochem. Photobiol., A, 2008, 199, 224–229 CrossRef CAS PubMed
- X. F. Zhou, C. Hu, X. X. Hu, T. W. Peng and J. H. Qu, J. Phys. Chem. C, 2010, 114, 2746–2750 CAS
- P. Wang, B. B. Huang, Z. Z. Lou, X. Y. Zhang, X. Y. Qin, Y. Dai, Z. K. Zheng and X. N. Wang, Chem.–Eur. J., 2010, 16, 538–544 CrossRef CAS PubMed
- C. Hu, T. W. Peng, X. X. Hu, Y. L. Nie, X. F. Zhou, J. H. Qu and H. He, J. Am. Chem. Soc., 2010, 132, 857–862 CrossRef CAS PubMed
- P. Wang, B. B. Huang, X. Y. Zhang, X. Y. Qin, H. Jin, Y. Dai, Z. Y. Wang, J. Y. Wei, J. Zhan, S. Y. Wang, J. P. Wang and M. H. Whangbo, Chem.–Eur. J., 2009, 15, 1821–1824 CrossRef CAS PubMed
- P. Wang, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai and M. H. Whangbo, Inorg. Chem., 2009, 48, 10697–10702 CrossRef CAS PubMed
- P. Wang, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai, J. Y. Wei and M. H. Whangbo, Angew. Chem., Int. Ed., 2008, 47, 7931–7933 CrossRef CAS PubMed
- Y. X. Tang, Z. L. Jiang, J. Y. Deng, D. G. Gong, Y. K. Lai, H. T. Tay, I. T. K. Joo, T. H. Lau, Z. L. Dong and Z. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 438–446 CAS
- Y. X. Tang, V. P. Subramaniam, T. H. Lau, Y. K. Lai, D. G. Gong, P. D. Kanhere, Y. H. Cheng, Z. Chen and Z. L. Dong, Appl. Catal., B, 2011, 106, 577–585 CrossRef CAS PubMed
- Y. Tang, Z. Jiang, G. Xing, A. Li, P. D. Kanhere, Y. Zhang, T. C. Sum, S. Li, X. Chen, Z. Dong and Z. Chen, Adv. Funct. Mater., 2013, 23, 2932–2940 CrossRef CAS PubMed
- P. Wang, Y. Tang, Z. Dong, Z. Chen and T.-T. Lim, J. Mater. Chem. A, 2013, 1, 4718–4727 CAS
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926–5940 RSC
- Z. H. Chen, I. Belharouak, Y. K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23, 959–969 CrossRef CAS PubMed
- Y. Tang, X. Rui, Y. Zhang, T. M. Lim, Z. Dong, H. H. Hng, X. Chen, Q. Yan and Z. Chen, J. Mater. Chem. A, 2013, 1, 82–88 CAS
- G. B. Xu, W. Li, L. W. Yang, X. L. Wei, J. W. Ding, J. X. Zhong and P. K. Chu, J. Power Sources, 2015, 276, 247–254 CrossRef CAS PubMed
- H. Yang, Z. Liu, B. K. Chandran, J. Deng, J. Yu, D. Qi, W. Li, Y. Tang, C. Zhang and X. Chen, Adv. Mater., 2015 DOI:10.1002/adma.201502484
- L. Kavan, M. Gratzel, J. Rathousky and A. Zukalb, J. Electrochem. Soc., 1996, 143, 394–400 CrossRef CAS PubMed
- J. Li, Z. Tang and Z. Zhang, Electrochem. Commun., 2005, 7, 62–67 CrossRef CAS PubMed
- J. Li, Z. Tang and Z. Zhang, Chem. Mater., 2005, 17, 5848–5855 CrossRef CAS
- B. L. Wang, Q. Chen, J. Hu, H. Li, Y. F. Hu and L. M. Peng, Chem. Phys. Lett., 2005, 406, 95–100 CrossRef CAS PubMed
- J. Li, W. Wan, F. Zhu, Q. Li, H. Zhou, J. Li and D. Xu, Chem. Commun., 2012, 48, 389–391 RSC
- Y. Y. Zhang, Y. X. Tang, S. Y. Yin, Z. Y. Zeng, H. Zhang, C. M. Li, Z. L. Dong, Z. Chen and X. D. Chen, Nanoscale, 2011, 3, 4074–4077 RSC
- F. Wu, Z. Wang, X. Li and H. Guo, J. Mater. Chem., 2011, 21, 12675–12681 RSC
- M. Wei, K. Wei, M. Ichihara and H. Zhou, Electrochem. Commun., 2008, 10, 1164–1167 CrossRef CAS PubMed
- A. G. Dylla, G. Henkelman and K. J. Stevenson, Acc. Chem. Res., 2013, 46, 1104–1112 CrossRef CAS PubMed
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, J. Power Sources, 2005, 146, 501–506 CrossRef CAS PubMed
- A. R. Armstrong, G. Armstrong, J. Canales, R. García and P. G. Bruce, Adv. Mater., 2005, 17, 862–865 CrossRef CAS PubMed
- G. Armstrong, A. R. Armstrong, P. G. Bruce, P. Reale and B. Scrosati, Adv. Mater., 2006, 18, 2597–2600 CrossRef CAS PubMed
- G. Armstrong, A. R. Armstrong, J. Canales and P. G. Bruce, Chem. Commun., 2005, 2454–2456 RSC
- G. Armstrong, A. R. Armstrong, J. S. Canales and P. G. Bruce, Electrochem. Solid-State Lett., 2006, 9, A139–A143 CrossRef CAS PubMed
- S. H. Liu, H. P. Jia, L. Han, J. L. Wang, P. F. Gao, D. D. Xu, J. Yang and S. N. Che, Adv. Mater., 2012, 24, 3201–3204 CrossRef CAS PubMed
- B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef PubMed
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed
- M. Lv, D. Zheng, M. Ye, J. Xiao, W. Guo, Y. Lai, L. Sun, C. Lin and J. Zuo, Energy Environ. Sci., 2013, 6, 1615–1622 CAS
- Y. Lai, Z. Lin, D. Zheng, L. Chi, R. Du and C. Lin, Electrochim. Acta, 2012, 79, 175–181 CrossRef CAS PubMed
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef CAS PubMed
- P. Roy, D. Kim, K. Lee, E. Spiecker and P. Schmuki, Nanoscale, 2010, 2, 45–59 RSC
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed
- M. Grätzel and J. R. Durrant, in Clean Electricity from Photovoltaics, 2014, pp. 413–452 Search PubMed
- Z. Tebby, O. Babot, D. Michau, L. Hirsch, L. Carlos and T. Toupance, J. Photochem. Photobiol., A, 2009, 205, 70–76 CrossRef CAS PubMed
- C. Magne, F. Dufour, F. Labat, G. Lancel, O. Durupthy, S. Cassaignon and T. Pauporté, J. Photochem. Photobiol., A, 2012, 232, 22–31 CrossRef CAS PubMed
- O. K. Varghese, M. Paulose and C. A. Grimes, Nat. Nanotechnol., 2009, 4, 592–597 CrossRef CAS PubMed
- C. Xu, J. Wu, U. V. Desai and D. Gao, Nano Lett., 2012, 12, 2420–2424 CrossRef CAS PubMed
- P. Docampo, S. Guldin, T. Leijtens, N. K. Noel, U. Steiner and H. J. Snaith, Adv. Mater., 2014, 26, 4013–4030 CrossRef CAS PubMed
- Y. Liu, R. Che, G. Chen, J. Fan, Z. Sun, Z. Wu, M. Wang, B. Li, J. Wei, Y. Wei, G. Wang, G. Guan, A. A. Elzatahry, A. A. Bagabas, A. M. Al-Enizi, Y. Deng, H. Peng and D. Zhao, Sci. Adv., 2015, 1, e1500166 Search PubMed
- N. Tétreault, É. Arsenault, L.-P. Heiniger, N. Soheilnia, J. Brillet, T. Moehl, S. Zakeeruddin, G. A. Ozin and M. Grätzel, Nano Lett., 2011, 11, 4579–4584 CrossRef PubMed
- Science News, Science, 2013, 342, 1438–1439 Search PubMed.
- N.-G. Park, Mater. Today, 2015, 18, 65–72 CrossRef CAS PubMed
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
- H.-S. Kim, C.-R. Lee, J.-H. Im, K.-B. Lee, T. Moehl, A. Marchioro, S.-J. Moon, R. Humphry-Baker, J.-H. Yum, J. E. Moser, M. Grätzel and N.-G. Park, Sci. Rep., 2012, 2, 591 Search PubMed
- W. A. Laban and L. Etgar, Energy Environ. Sci., 2013, 6, 3249–3253 CAS
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed
- N. J. Jeon, J. H. Noh, W. S. Yang, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Nature, 2015, 517, 476–480 CrossRef CAS PubMed
- http://www.nrel.gov/ncpv/images/efficiency_chart.jpg
- M.-E. Ragoussi and T. Torres, Chem. Commun., 2015, 51, 3957–3972 RSC
- D. Kowalski, D. Kim and P. Schmuki, Nano Today, 2013, 8, 235–264 CrossRef CAS PubMed
- K. Lee, A. Mazare and P. Schmuki, Chem. Rev., 2014, 114, 9385–9454 CrossRef CAS PubMed
- Z. Tebby, O. Babot, T. Toupance, D. H. Park, G. Campet and M. H. Delville, Chem. Mater., 2008, 20, 7260–7267 CrossRef CAS
- A. Ghicov, H. Tsuchiya, R. Hahn, J. M. Macak, A. G. Munoz and P. Schmuki, Electrochem. Commun., 2006, 8, 528–532 CrossRef CAS PubMed
- A. Ghicov, S. P. Alba, J. M. Macak and P. Schmuki, Small, 2008, 4, 1063–1066 CrossRef CAS PubMed
- Y. Sanehira, S. Uchida, T. Kubo and H. Segawa, Electrochemistry, 2008, 76, 150–153 CrossRef CAS PubMed
- S. Berger, A. Ghicov, Y. C. Nah and P. Schmuki, Langmuir, 2009, 25, 4841–4844 CrossRef CAS PubMed
- P. Roy, S. Berger and P. Schmuki, Angew. Chem., Int. Ed., 2011, 50, 2904–2939 CrossRef CAS PubMed
- L. Yao and J. He, Prog. Mater. Sci., 2014, 61, 94–143 CrossRef PubMed
- D. Lee, M. F. Rubner and R. E. Cohen, Nano Lett., 2006, 6, 2305–2312 CrossRef CAS PubMed
- W. Barthlott and C. Neinhuis, Planta, 1997, 202, 1–8 CrossRef CAS
- S. Nishimoto and B. Bhushan, RSC Adv., 2013, 3, 671–690 RSC
- X. Ding, S. Zhou, G. Gu and L. Wu, J. Mater. Chem., 2011, 21, 6161–6164 RSC
- T. Kamegawa, Y. Shimizu and H. Yamashita, Adv. Mater., 2012, 24, 3697–3700 CrossRef CAS PubMed
- Y. Lu, S. Sathasivam, J. Song, C. R. Crick, C. J. Carmalt and I. P. Parkin, Science, 2015, 347, 1132–1135 CrossRef CAS PubMed
- J. Huang, Y. Lai, L. Wang, S. Li, M. Ge, K. Zhang, H. Fuchs and L. Chi, J. Mater. Chem. A, 2014, 2, 18531–18538 CAS
- Y. Sawai, S. Nishimoto, Y. Kameshima, E. Fujii and M. Miyake, Langmuir, 2013, 29, 6784–6789 CrossRef CAS PubMed
- Y. Lai, Y. Tang, J. Huang, H. Wang, H. Li, D. Gong, X. Ji, J. Gong, C. Lin, L. Sun and Z. Chen, Soft Matter, 2011, 7, 6313–6319 RSC
- Y. Lai, H. Zhou, Z. Zhang, Y. Tang, J. W. C. Ho, J. Huang, Q. Tay, K. Zhang, Z. Chen and B. P. Binks, Part. Part. Syst. Charact., 2015, 32, 355–363 CrossRef CAS PubMed
- Y. Lai, Y. Tang, J. Gong, D. Gong, L. Chi, C. Lin and Z. Chen, J. Mater. Chem., 2012, 22, 7420–7426 RSC
- H. Li, J. Wang, L. Yang and Y. Song, Adv. Funct. Mater., 2008, 18, 3258–3264 CrossRef CAS PubMed
- Q. Zhu, F. Tao and Q. Pan, ACS Appl. Mater. Interfaces, 2010, 2, 3141–3146 CAS
- L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., 2004, 116, 2046–2048 CrossRef PubMed
- B. Wang, W. Liang, Z. Guo and W. Liu, Chem. Soc. Rev., 2015, 44, 336–361 RSC
- J. Yuan, X. Liu, O. Akbulut, J. Hu, S. L. Suib, J. Kong and F. Stellacci, Nat. Nanotechnol., 2008, 3, 332–336 CrossRef CAS PubMed
- X. Gui, J. Wei, K. Wang, A. Cao, H. Zhu, Y. Jia, Q. Shu and D. Wu, Adv. Mater., 2010, 22, 617–621 CrossRef CAS PubMed
- L. Li, Z. Liu, Q. Zhang, C. Meng, T. Zhang and J. Zhai, J. Mater. Chem. A, 2015, 3, 1279–1286 CAS
- C. Gao, Z. Sun, K. Li, Y. Chen, Y. Cao, S. Zhang and L. Feng, Energy Environ. Sci., 2013, 6, 1147–1151 CAS
- L. Zhang, Y. Zhong, D. Cha and P. Wang, Sci. Rep., 2013, 3, 2326 Search PubMed
- Y.-K. Lai, Y.-X. Tang, J.-Y. Huang, F. Pan, Z. Chen, K.-Q. Zhang, H. Fuchs and L.-F. Chi, Sci. Rep., 2013, 3, 3009 Search PubMed
- J. Y. Huang, S. H. Li, M. Z. Ge, L. N. Wang, T. L. Xing, G. Q. Chen, X. F. Liu, S. S. Al-Deyab, K. Q. Zhang, T. Chen and Y. K. Lai, J. Mater. Chem. A, 2015, 3, 2825–2832 CAS
- H. Li, Y. Lai, J. Huang, Y. Tang, L. Yang, Z. Chen, K. Zhang, X. Wang and L. P. Tan, J. Mater. Chem. B, 2015, 3, 342–347 RSC
- S. Wu, Z. Weng, X. Liu, K. W. K. Yeung and P. K. Chu, Adv. Funct. Mater., 2014, 24, 5464–5481 CrossRef CAS PubMed
- K. Huo, B. Gao, J. Fu, L. Zhao and P. K. Chu, RSC Adv., 2014, 4, 17300–17324 RSC
- G. Petroffe, C. Wang, X. Sallenave, G. Sini, F. Goubard and S. Peralta, J. Mater. Chem. A, 2015, 3, 11533–11542 CAS
- J. Y. Huang, Y. K. Lai, F. Pan, L. Yang, H. Wang, K. Q. Zhang, H. Fuchs and L. F. Chi, Small, 2014, 10, 4865–4873 CrossRef CAS PubMed
- Y. K. Lai, Y. C. Chen, Y. X. Tang, D. G. Gong, Z. Chen and C. J. Lin, Electrochem. Commun., 2009, 11, 2268–2271 CrossRef CAS PubMed
- Y. K. Lai, C. J. Lin, J. Y. Huang, H. F. Zhuang, L. Sun and T. Nguyen, Langmuir, 2008, 24, 3867–3873 CrossRef CAS PubMed
- Y. K. Lai, X. F. Gao, H. F. Zhuang, J. Y. Huang, C. J. Lin and L. Jiang, Adv. Mater., 2009, 21, 3799–3803 CrossRef CAS PubMed
- K. Liu, M. Cao, A. Fujishima and L. Jiang, Chem. Rev., 2014, 114, 10044–10094 CrossRef CAS PubMed
- S. Li, J. Huang, M. Ge, C. Cao, S. Deng, S. Zhang, G. Chen, K. Zhang, S. S. Al-Deyab and Y. Lai, Adv. Mater. Interfaces, 2015, 2, 1500220 Search PubMed
- H. Wang, Y. K. Lai, R. Y. Zheng, Y. Bian, K. Q. Zhang and C. J. Lin, Int. J. Nanomed., 2015, 10, 3887 CrossRef PubMed
- Y. Koc, A. J. de Mello, G. McHale, M. I. Newton, P. Roach and N. J. Shirtcliffe, Lab Chip, 2008, 8, 582–586 RSC
- X. Hou, X. Wang, Q. Zhu, J. Bao, C. Mao, L. Jiang and J. Shen, Colloids Surf., B, 2010, 80, 247–250 CrossRef CAS PubMed
- Y. Lai, F. Pan, C. Xu, H. Fuchs and L. Chi, Adv. Mater., 2013, 25, 1682–1686 CrossRef CAS PubMed
- T. Ishizaki, N. Saito and O. Takai, Langmuir, 2010, 26, 8147–8154 CrossRef CAS PubMed
- X. Zhang, L. Wang and E. Levanen, RSC Adv., 2013, 3, 12003–12020 RSC
- Y. Yang, Y. Lai, Q. Zhang, K. Wu, L. Zhang, C. Lin and P. Tang, Colloids Surf., B, 2010, 79, 309–313 CrossRef CAS PubMed
- Q. Huang, Y. Yang, R. Hu, C. Lin, L. Sun and E. A. Vogler, Colloids Surf., B, 2015, 125, 134–141 CrossRef CAS PubMed
- V. K. Truong, H. K. Webb, E. Fadeeva, B. N. Chichkov, A. H. F. Wu, R. Lamb, J. Y. Wang, R. J. Crawford and E. P. Ivanova, Biofouling, 2012, 28, 539–550 CrossRef CAS PubMed
- P. Tang, W. Zhang, Y. Wang, B. Zhang, H. Wang, C. Lin and L. Zhang, J. Nanomater., 2011, 2011, 178921 Search PubMed
- Y. Lai, L. Lin, F. Pan, J. Huang, R. Song, Y. Huang, C. Lin, H. Fuchs and L. Chi, Small, 2013, 9, 2945–2953 CrossRef CAS PubMed
- Y. Lai, C. Lin, H. Wang, J. Huang, H. Zhuang and L. Sun, Electrochem. Commun., 2008, 10, 387–391 CrossRef CAS PubMed
- Y.-X. Huang, Y.-K. Lai, L.-X. Lin, L. Sun and C.-J. Lin, Acta Phys.-Chim. Sin., 2010, 26, 2057–2060 CAS
- Y. Lai, J. Huang, J. Gong, Y. Huang, C. Wang, Z. Chen and C. Lin, J. Electrochem. Soc., 2009, 156, D480–D484 CrossRef CAS PubMed
- Y. Huang, L. Sun, K. Xie, Y. Lai, B. Liu, B. Ren and C. Lin, J. Raman Spectrosc., 2011, 42, 986–991 CrossRef CAS PubMed
- Y. Lai, Y. Huang, H. Wang, J. Huang, Z. Chen and C. Lin, Colloids Surf., B, 2010, 76, 117–122 CrossRef CAS PubMed
- S. Bekou and D. Mattia, Curr. Opin. Colloid Interface Sci., 2011, 16, 259–265 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2015 |