
Asymmetric and symmetric solid-state supercapacitors based on 3D interconnected polyaniline–carbon nanotube framework
Abstract
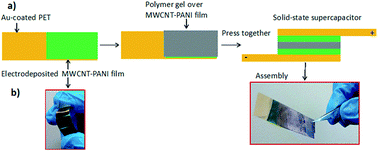
In this work, we demonstrate multiwalled carbon nanotube (MWCNT)-assisted polyaniline (PANI) thin films to improve supercapacitor performance. The thin films of PANI have been potentiostatically deposited onto gold coated PET sheet with the assistance of MWCNT. The assistance of MWCNT results in the formation of unique nanostructured PANI framework which provides better accessibility for supercapacitive behavior. The solid-state supercapacitor has been made using two slightly separated thin films of MWCNT-assisted PANI (MWCNT-PANI) by H3PO4–polyvinyl alcohol (H3PO4–PVA) gel electrolyte. The asymmetric solid-state supercapacitor (ASS) and symmetric solid-state supercapacitor (SSS) devices exhibit the remarkable area-specific capacitance of 23.1 mF cm−2 (660 F cm−3) and 8.3 mF cm−2 (119 F cm−3), respectively. The ASS and SSS devices have the energy density 2.7 mW h cm−2 and 0.95 mW h cm−2 while maintaining the power density of 337 mW cm−2 and 263 mW cm−2, and excellent cyclic stability. The electrochemical properties of MWCNT-PANI films have also been investigated in aqueous H2SO4 and organic tetraethyl ammonium tetrafluoroborate electrolytes in half cell configuration.
 Please wait while we load your content...
Please wait while we load your content...

