
Glutaraldehyde sandwiched amino functionalized polymer based aptasensor for the determination and quantification of chloramphenicol
Abstract
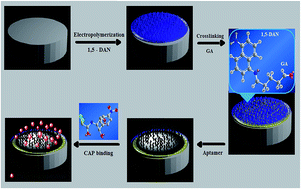
In the present work, the crosslinking property of glutaraldehyde (GA) is exploited to construct a 1,5-diaminonaphthalene (1,5-DAN) modified aptamer based sensor for the determination and quantification of chloramphenicol (CAP). The sensor was fabricated by electro-polymerizing 1,5-DAN on the edge plane pyrolytic graphite (EPPG) followed by immobilization of CAP selective aptamer on the amino functionalized polymer modified EPPG surface using GA, which resulted in covalent linkage with the –NH2 groups of the aptamer as well as those present on the polymerized electrode surface. Resulting GA sandwiched aptasensor leads to a tremendous improvement in the stability and sensitivity of the sensor in comparison to one fabricated without crosslinking. Characterization of the modification protocol was done using scanning electron microscopy (SEM), quartz crystal microbalance (QCM) analysis, and electrochemical impedance spectroscopy (EIS). Under optimized parameters, CAP was detected in a linear working range of 50–500 fM with the detection limit and sensitivity of 11 fM (n = 3, R.S.D = 0.15%) and 0.023 μA fM−1 respectively. The practical utility of the developed aptasensor has also been demonstrated by analysing CAP in complex matrix like urine, commercially available pharmaceutical samples and the Rosetta cells supernatant. The observed results suggests that the present electrochemical aptasensor is suitable for the detection of CAP in medicines, diagnosis of chloramphenicol resistant bacteria and virus, as well as for checking its adulteration in food and other dietary supplements.
 Please wait while we load your content...
Please wait while we load your content...

