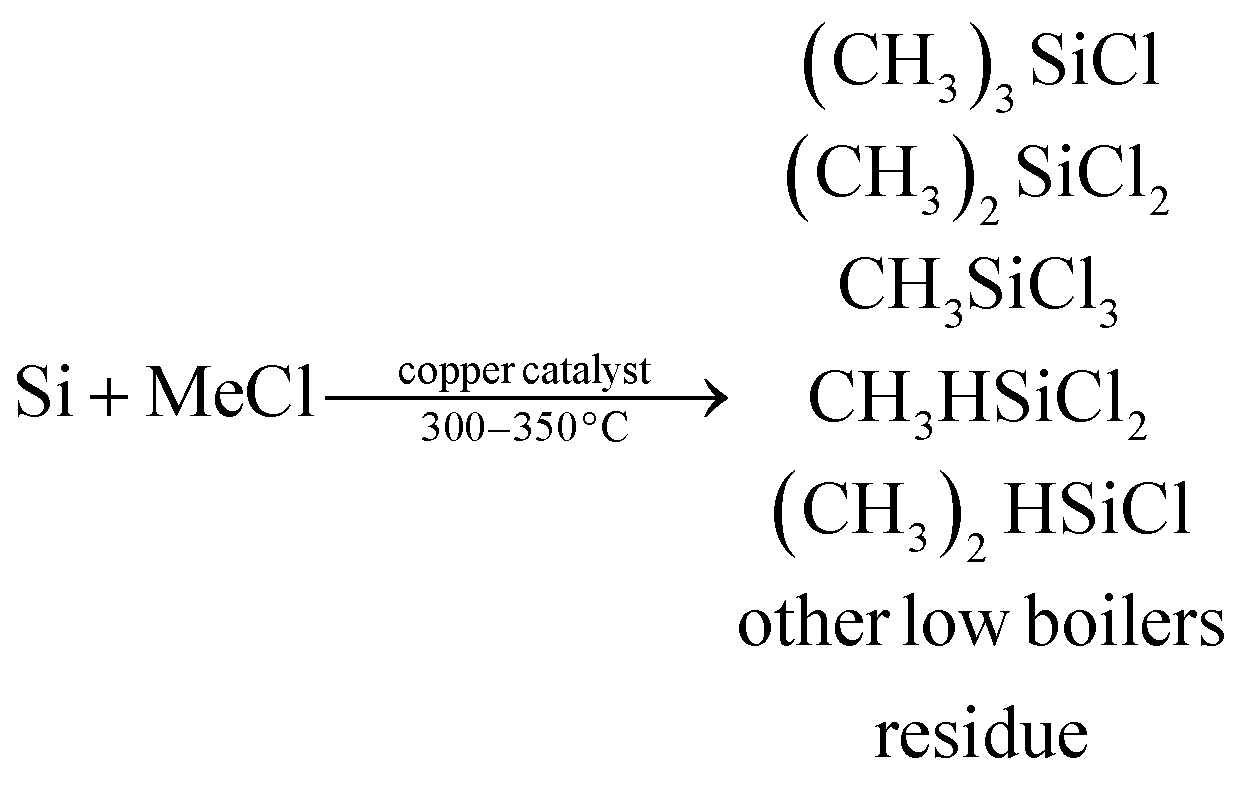DOI:
10.1039/C5RA10999J
(Paper)
RSC Adv., 2015,
5, 73011-73019
Controllable wet synthesis of multicomponent copper-based catalysts for Rochow reaction†
Received
10th June 2015
, Accepted 21st August 2015
First published on 21st August 2015
Abstract
This work aims to provide a facile, low-cost and scalable method for the preparation of multicomponent Cu–Cu2O–CuO catalysts, which are of high interest to the organosilane industry. A series of submicrometer-sized and Cu-based catalysts containing CuO, Cu2O and Cu, or some combination of them, were synthesized by a simple low-temperature wet chemical method using CuSO4·5H2O as the precursor and N2H4·H2O as a reducing agent. The samples were characterized by X-ray diffraction, thermogravimetric analysis, temperature-programmed reduction, X-ray photoelectron spectroscopy, transmission electron microscopy, and scanning electron microscopy techniques. It was observed that the composition of the samples could be tailored by varying the amount of reducing agent at a given reaction temperature and time. These catalysts were then tested in the Rochow reaction, using silicon powder and methyl chloride (MeCl) as reactants to produce dimethyldichlorosilane (M2), which is the most important organosilane monomer in the industry. Compared with bare CuO and Cu particles, the ternary CuO–Cu2O–Cu catalyst displayed much improved M2 selectivity and Si conversion, which can be attributed to the smaller copper particle size and the synergistic effect among the different components in the CuO–Cu2O–Cu catalyst. This catalyst preparation method is expected to yield efficient and low-cost copper catalysts for the organosilane industry.
1. Introduction
The Rochow reaction, which was discovered in the 1940s,1 is still the most economical route to produce silicone monomers in the organosilane industry. In this reaction, gaseous methyl chloride (MeCl) reacts with silicon (Si) in the presence of a Cu-based catalyst and a trace amount of promoter as follows:2,3| |
 | (1) |
Among the products from the above Rochow reaction, dimethyldichlorosilane ((CH3)2SiCl2, M2) is the most important monomer used for silicon rubber production in the organosilane industry, and thus, a high M2 yield is highly desired. Besides the accurate control of reaction conditions and the selection of proper reactors, the development of highly efficient catalytic systems is crucial for high M2 selectivity and Si conversion. As reported, the primary catalysts used in the Rochow reaction are Cu-based catalysts including metallic Cu,1 CuCl,4 Cu2O,5 CuO,6 Cu–Si alloy7 and Cu–Cu2O–CuO,8 together with some catalyst promoters.2,6,9–11 In recent years, our group has developed a number of cupreous and copper oxide structures, such as flower-like ZnO grown on urchin-like CuO microspheres,12 porous cubic Cu microparticles,13 mesoporous Cu2O microspheres,5 shape-controlled Cu2O microparticles,14 dandelion-like CuO microspheres,15 flower-like CuO microspheres,16 and CuCl microcrystals with different morphologies17 etc., for M2 synthesis, and found that the particle morphology and size, as well as the catalyst composition, impact on the catalytic properties significantly.
Among the various Cu-based catalysts, the multicomponent Cu-based catalysts containing Cu, Cu2O, and CuO are of great interest due to their superior catalytic performances in the Rochow reaction. For instance, Khitouni et al. prepared such catalysts through a high-energy mechanical milling process, using Cu, Cu2O, and CuO as the precursors.18 Although their method is simple, it still lacks effective control of the solid reaction between Cu, Cu2O, and CuO during the mechanical milling process. Therefore, to meet industrial application, more effective methods for preparing multicomponent Cu-based catalysts should be developed. More recently, we found that the multicomponent Cu–Cu2O–CuO catalyst can also be synthesized by the partial reduction of CuO nanoparticles in H2/N2 gas19 or by the controlled oxidation of copper flakes in O2/N2 gas.20 These methods, however, still suffer from low yields and the use of high-cost equipment together with intensive energy consumption (high temperature), which are troublesome and thus limit their further application. On the other hand, we have noticed that there are scarce reports concerning the preparation of multicomponent Cu-based catalysts via wet chemical approaches.
Herein, we report a facile and low cost preparation of the ternary CuO–Cu2O–Cu catalyst by a wet chemical method with obvious advantages, such as mild reaction conditions (70 °C), large-scale production capability and easy operation. Most importantly, the composition of the samples could be tailored by simply varying the amount of the reducing agent N2H4·H2O. The prepared ternary CuO–Cu2O–Cu catalyst exhibited highly improved M2 selectivity and Si conversion in the Rochow reaction in comparison to those of sole CuO and Cu microparticles, demonstrating the importance of the synergistic catalytic effect among the components. The resulting ternary CuO–Cu2O–Cu catalyst using the present method is a promising catalyst for the industrial Rochow reaction.
2. Experimental
2.1. Material synthesis
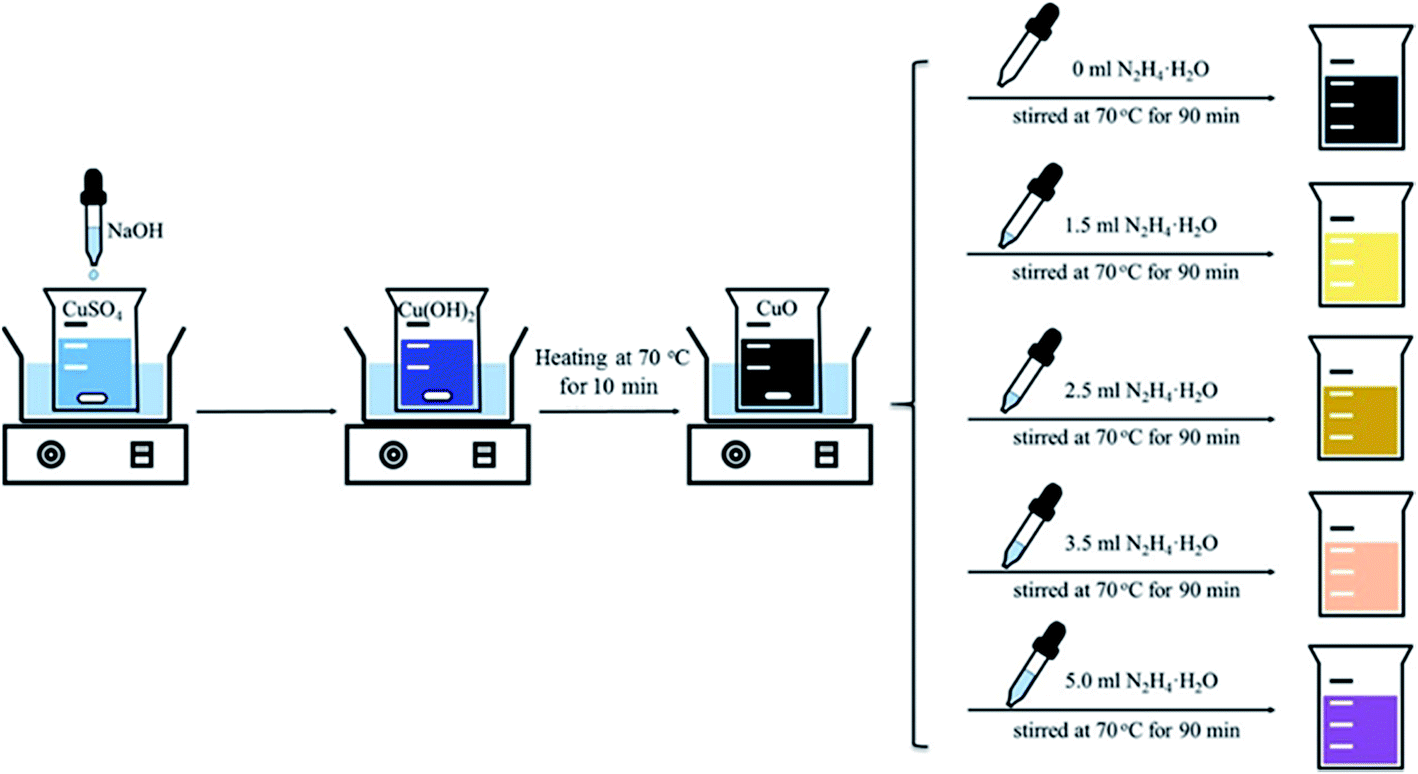
The samples were synthesized using a wet chemical reduction method as shown in Fig. 1. In a typical preparation, 37.5 g of copper(II) sulfate pentahydrate (CuSO4·5H2O, A.R., Sinopharm Chemical Reagent Co., Ltd., China) was dissolved in 300.0 mL of deionized water to get a clear solution with a copper concentration of 0.5 M. Upon heating to 70 °C under magnetic stirring, a sodium hydroxide (NaOH, A.R., Sinopharm Chemical Reagent Co., Ltd.) aqueous solution (6 M) was added dropwise until the pH value of the solution reached 7.5. The obtained mixed gel was maintained for 10 min at this temperature to give a black precipitate. Subsequently, desired amounts of hydrazine hydrate (N2H4·H2O), used as the reductant, were introduced dropwise. After being stirred for another 90 min, the reaction mixture was cooled down to room temperature naturally. The resulting solid was collected by filtration, washed with deionized water and ethanol several times, and finally dried in a vacuum at 60 °C for 8 h. The synthesis was carried out by fixing the Cu amount at 9.5 g while varying the amount of N2H4·H2O added. The sample compositions were determined by XRD analysis, and the prepared samples are thus denoted as CuO, CuO–Cu2O, CuO–Cu2O–Cu, Cu2O–Cu and Cu according to their measured compositions when the amount of N2H4·H2O was 0, 1.5, 2.5, 3.5 and 5.0 mL, respectively.
 |
| | Fig. 1 Schematic illustration of the preparation process. | |
2.2. Characterization
X-ray diffraction (XRD) patterns were recorded on a PANalytica X’Pert PRO MPD diffractometer using the Kα radiation of Cu (λ = 1.5418 Å) and checked with the card number of the Joint Committee on Powder Diffraction Standards (JCPDS). The crystallite size was calculated using the Scherrer equation, in which the shape factor of K is 0.90. Moreover, the 2θ value of instrumental broadening is 0.06, which was subtracted in the test process. The microscopic feature of the samples was observed by field-emission scanning electron microscopy (SEM) (JSM-7001F, JEOL, Tokyo, Japan) with energy-dispersive spectroscopy (EDS) (INCA X-MAX, JEOL, Oxford, England) and transmission electron microscopy (TEM) (JEM-2010F, JEOL, Tokyo, Japan). The specific surface area was determined according to the Brunauer–Emmett–Teller (BET) method in the relative pressure range of 0.05–0.2. Thermal gravimetric (TG) analysis was carried out on an EXSTAR TG/DTA 6300 (Seiko Instruments, Japan) apparatus with a heating rate of 10 °C min−1 in air (200 mL min−1). H2-temperature programmed reduction (H2-TPR) measurements were carried out on an automated chemisorption analyzer (ChemBET pulsar TPR/TPD, Quantachrome). Upon the loading of about 0.10 g of the sample into a quartz U-tube, the sample was degassed at 150 °C for 30 min under helium. When the temperature decreased to 30 °C, the gas was changed to 9.9% O2/He. Finally, the sample was heated from 30 to 900 °C at a rate of 10 °C min−1 in 9.9% O2/He gas with a gas flow of 30 mL min−1. X-ray photoelectron spectroscopy (XPS) analysis was carried out on an ESCALAB 250Xi apparatus from Thermo Scientific Corporation using AlKα X-ray radiation.
2.3. Catalytic measurement
The evaluation of the catalyst was carried out on a homemade lab fixed-bed reactor. 10.0 g Si powder (20–50 mesh, provided by Jiangsu Hongda New Material Co., Ltd.) and 1.0 g of prepared catalyst, together with 0.10 g of zinc (Zn, A.R., Sinopharm Chemical Reagent Co., Ltd.) which was used as a promoter, were mixed homogeneously to form a contact mass, which was loaded in the glass reactor. The reactor system was purged with high purity N2 for 0.5 h, followed by heating to 325 °C within 1 h under a N2 flow rate of 25 mL min−1. Subsequently, the gas was switched to MeCl gas with a flow rate of 25 mL min−1 to react with Si at 325 °C. After 24 h, the reaction was ceased. The gas products were cooled with a circulator bath controlled at 5 °C by a programmable thermal circulator (GDH series, Ningbo xinzhi biological technology Co., Ltd.) and then identified by gas chromatography mass spectrometry (GC-MS) (QP2010, SHIMADZU) to be composed of methyltrichlorosilane (CH3SiCl3, M1), dimethyldichlorosilane ((CH3)2SiCl2, M2), trimethylchlorosilane ((CH3)3SiCl, M3), methyldichlorosilane (CH3SiHCl2, M1H), dimethylchlorosilane ((CH3)2SiHCl, M2H), low boiler (LB) and high boiler (HB). They were quantitatively analyzed on a gas chromatograph (Agilent Technologies 7890A, Thermal conductivity detector, KB-201 column). Following the previously reported method,13 the selectivity of the products was calculated by the peak area ratio (in percentage) and the Si conversion was obtained from the difference of contact masses (before and after the reaction), as expressed as follows:| |
 | (2) |
3. Results and discussion
3.1. Characterizations of Cu-based catalysts
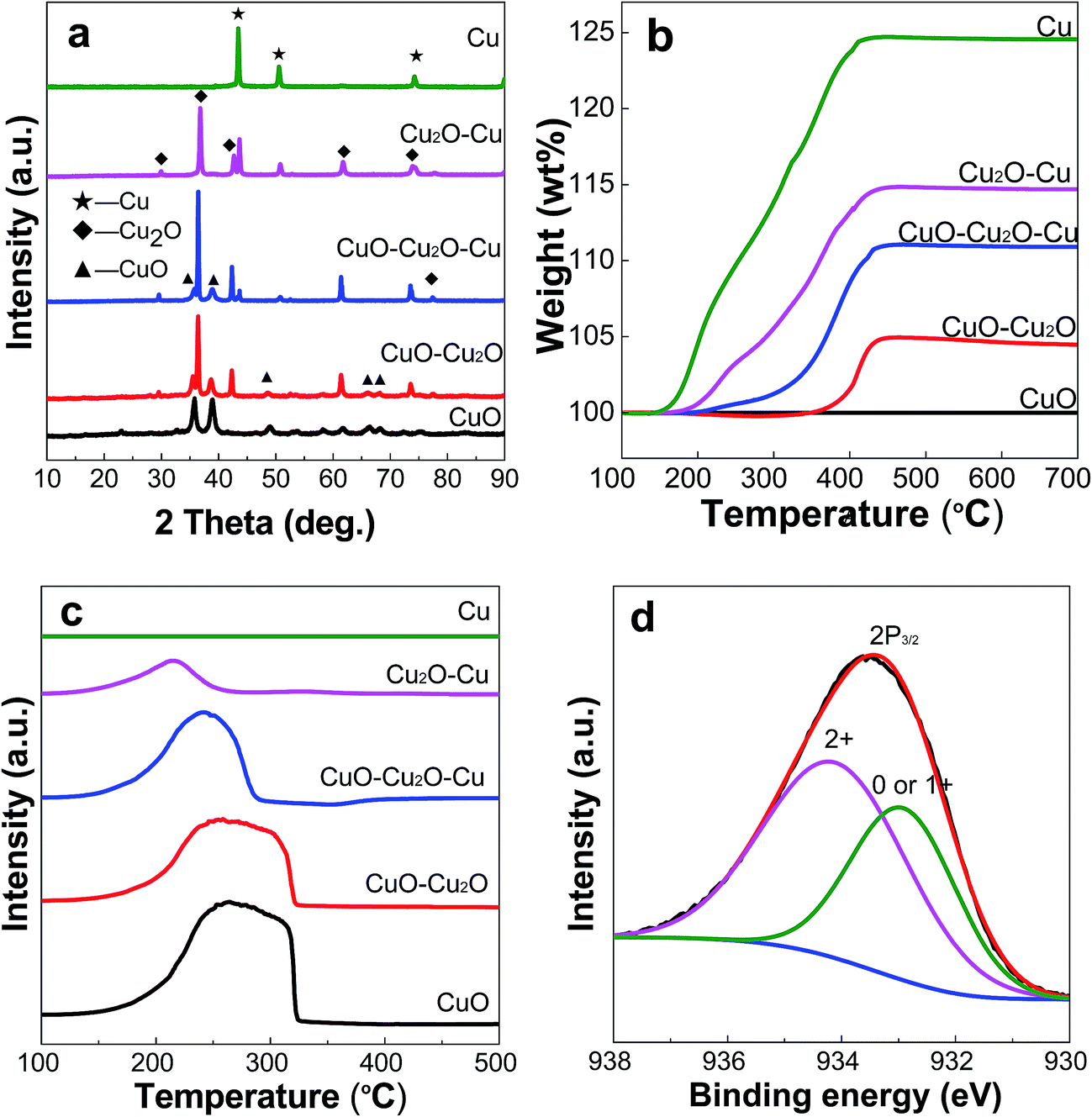
Fig. 2a shows the XRD patterns of all the samples, which have high diffraction intensities, indicating their good crystallinity. In the absence of N2H4·H2O, diffraction peaks at 2θ values of 33.5°, 35.5°, 38.2°, 48.7°, 54.2°, 58.3°, 62.5°, 66.4°, 68.2°, 73.4°, and 75.6° are observed for the product, which correspond respectively to the lattice planes of (110), (−111), (111), (−202), (020), (202), (−113), (−311), (220), (311), and (004) of monoclinic CuO (JCPDS No. 03-065-2309). Compared to those of CuO, the sample prepared with 1.5 mL of N2H4·H2O has additional diffraction peaks at 2θ = 36.7° and 42.7°, which are attributed to the (111) and (200) planes of cubic Cu2O (JCPDS No. 00-001-1142), suggesting the formation of a bicomponent system of CuO and Cu2O. By increasing the amount of N2H4·H2O to 2.5 mL, in addition to the above CuO–Cu2O diffraction peaks, new peaks at 2θ values of 43.3°, 50.5°, and 74.2° appeared for the sample, which are assigned to the lattice planes of (111), (200), and (220) of cubic Cu (JCPDS No. 01-070-3039), revealing that the sample contains CuO, Cu2O and Cu. With a further increase of the N2H4·H2O amount up to 3.5 mL or 5.0 mL, the XRD patterns show that the obtained samples contain Cu2O and Cu or pure metallic Cu. The nanocrystal sizes of CuO, Cu2O, and Cu for each sample were calculated using the Scherrer formula based on the diffraction peaks at 2θ values of 35.5°, 36.7,°and 43.3°, respectively, and these data are compiled in Table 1. For bare CuO, the nanocrystal has a mean size of 18 nm, while in the samples of CuO–Cu2O and CuO–Cu2O–Cu, the nanocrystal size of CuO is decreased from 18 to 17 nm. On the other hand, the nanocrystal size of Cu2O in CuO–Cu2O is 45 nm, which is smaller than that in CuO–Cu2O–Cu (51 nm) but slightly larger than that in Cu2O–Cu (41 nm). In a difference from CuO and Cu2O, the nanocrystal size of Cu in CuO–Cu2O–Cu (35 nm), Cu2O–Cu (46 nm), and Cu (51 nm) increased progressively with the increasing amount of N2H4·H2O. We once partially reduced CuO nanoparticles in H2/N2 mixed gas with the purpose of obtaining multicomponent Cu-based catalysts, and also observed this trend in size change.19
 |
| | Fig. 2 XRD patterns (a), TG curves (b), and H2-TPR curves (c) of all the samples, and XPS spectra of the Cu 2p peak for the CuO–Cu2O–Cu sample (d). | |
Table 1 The physical chemistry parameters of all the samples
| Samples |
Cua (nm) |
Cu2Oa (nm) |
CuOa (nm) |
Weight increaseb (wt%) |
TMc (°C) |
STPRc |
SBETd (m2 g−1) |
| Calculated from the XRD patterns, standard errors: crystal size, 0.5 nm. Given by TG analysis. Obtained from H2-TPR curves using the integrated areas under the curves presented. Calculated with the BET method. |
| CuO |
— |
— |
18 |
— |
264.2 |
53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 460.8 460.8 |
15.3 |
| CuO–Cu2O |
— |
45 |
18 |
4.5 |
258.5 |
34482.3 |
10.2 |
| CuO–Cu2O–Cu |
35 |
51 |
17 |
11.2 |
241.2 |
23146.7 |
3.6 |
| Cu2O–Cu |
46 |
41 |
— |
17.1 |
214.5 |
9435.0 |
3.4 |
| Cu |
51 |
— |
— |
25.2 |
— |
— |
2.5 |
Fig. 2b shows the TG curves of all the samples measured in air. It is seen that there is no visible weight change for the CuO sample, suggesting the formation of a nearly pure CuO phase. The CuO–Cu2O sample shows a weight increase of 4.5 wt% over a wide temperature range (200–420 °C) which is derived from the oxidation of Cu2O to CuO. Thus, the estimated content of Cu2O in this sample is about 40.2 wt%. On the other hand, the CuO–Cu2O–Cu and Cu2O–Cu samples exhibit a 11.2 and 17.1 wt% weight increase, respectively, corresponding to the oxidation of Cu2O and Cu to CuO. In the case of Cu, the weight increase reaches 25.2 wt%, which is close to the theoretical value of the oxidation of Cu into CuO (25.0 wt%), confirming that it is metallic Cu.
Fig. 2c shows the H2-TPR curves of all the samples. The hydrogen consumption peak areas (STPR) and peak temperatures (TM) are summarized in Table 1. It is observed that with the decrease of CuO content in the samples, following the order of CuO, CuO–Cu2O, CuO–Cu2O–Cu, Cu2O–Cu and Cu, the reduction temperature and hydrogen consumption are also slightly decreased. For the reduced samples, the reduction peaks move slightly to a lower temperature region with an increase in the amount of reducing agent. Using the ratio of hydrogen consumption peak area to the mole number of O in CuO as a reference, and based on the above TG analysis, it can be calculated that the content of CuO in the CuO–Cu2O and CuO–Cu2O–Cu samples is about 59.4 and 15.7 wt%, respectively. For Cu2O–Cu, the content of Cu2O is 40.6 wt%. Therefore, the above results indicate that the contents of Cu, Cu2O, and CuO in the final products can be well tuned by adjusting the amount of N2H4·H2O in the preparation.
The high-resolution XPS spectra in Fig. 2d show the binding energies of Cu 2p3/2. The peaks at 934.2 eV are characteristic of Cu(II), whereas the ones at 932.9 eV are attributed to Cu (0) or Cu(I).21,22 Unfortunately, from the XPS data alone, it is difficult to distinguish clearly the Cu(0) and Cu(I) species due to the effects of crystal size and surface coverage on the binding energy.23 However, by combining them with the XRD results shown in Fig. 2a, we can judge whether the sample contains Cu (0) or Cu(I), or both of them. In other words, we can conclude that the sample actually contains CuO, Cu2O and Cu.
Fig. 3 shows the SEM images of all the samples. In agreement with the XRD results, the samples all appear to be well crystallized. Fig. 3a presents the SEM image of CuO, which reveals that the sample is made up of a large quantity of different sheet-like submicrometer structures. However, after the addition of a small amount of N2H4·H2O, the SEM image shows that the obtained CuO–Cu2O sample is composed of premature polyhedron structures, the surfaces of which are covered with a sheet-like substance (Fig. 3b), implying that the CuO crystallites are partially reduced to Cu2O and attached to the surface simultaneously during the growth of the polyhedron structures, which coincides with previous studies.24–26 A further increase in the amount of N2H4·H2O leads to obvious changes in the morphology of CuO–Cu2O–Cu (Fig. 3c), in which the sample consists of spheres, cubes, octahedra and other polyhedra in sizes ranging from approximately 0.05 to 2 μm. Fig. 3d displays the SEM image of Cu2O–Cu, in which the majority of the crystals have cubic and octahedral shapes, and their sizes are in the range of 100–500 nm. The Cu sample, as shown in Fig. 3e, exhibits a block-like shape with a size range of 0.1–3 μm.
 |
| | Fig. 3 SEM images of CuO (a), CuO–Cu2O (b), CuO–Cu2O–Cu (c), Cu2O–Cu (d), and Cu (e). | |
Fig. 4a presents the TEM image of the CuO–Cu2O–Cu sample, which consists of cubic, octahedral, spherical and other polyhedral crystals, in full agreement with the above SEM result (Fig. 3c). The high-resolution transmission electron microscope (HRTEM) image of CuO–Cu2O in Fig. 4b shows the presence of two different interplanar distances of 0.25 and 0.24 nm, corresponding to the (111) planes of CuO and Cu2O, respectively. Moreover, two sets of diffraction spots in the fast Fourier transform (FFT) patterns further verify the coexistence of the two structures. Similarly, the CuO–Cu2O–Cu sample (Fig. 4c) possesses three apparent lattice spacings, in which the one at 0.21 nm corresponds to the (111) plane of Cu, and the other two, indexed to the (111) planes of CuO and Cu2O, remain the same (0.25 and 0.24 nm) as CuO–Cu2O. In the case of Cu2O–Cu (Fig. 4d), two lattice plane distances of 0.24 and 0.21 nm are observed, which are characteristic of the (111) facets of Cu2O and CuO, respectively. These results prove that the prepared Cu-based samples are composed of polycrystals, while within each multi-component particle there are different Cu component crystals, supporting the successful synthesis of Cu-based catalysts with controllable composition by varying the amount of reducing agent using a wet chemical method.
 |
| | Fig. 4 TEM image of CuO–Cu2O–Cu (a), HRTEM images of CuO–Cu2O (top and bottom insets show the corresponding FFT pattern of CuO (−111) and Cu2O (111), respectively) (b), CuO–Cu2O–Cu (c), and Cu2O–Cu (d). | |
3.2. Reduction mechanism
The reactions involved in CuO synthesis and the reduction processes are shown in the following equations:| | |
CuSO4 + 2NaOH → Cu(OH)2 + Na2SO4
| (3) |
| | |
CuO + N2H4 → 2Cu2O + N2 + 2H2O
| (5) |
| | |
Cu2O + N2H4 → 2Cu + N2 + H2O
| (6) |
The chemical reaction between CuSO4 and NaOH yields cupric hydroxide (Cu(OH)2) (formula (3)), which is a metastable phase. Upon heating in a constant temperature (70 °C) water bath under magnetic stirring, it decomposes into more stable cupric oxide (CuO) (formula (4)),27 which has been also confirmed previously by Cudennec and Lecerf et al.28 Upon the addition of N2H4·H2O, which is employed as a reducing agent, CuO is reduced into Cu2O and Cu, accompanied with the production of N2 and H2O (formulas (5) and (6)).24,25 In addition, we also found that a trace of NH3 as a by-product will also be generated. The formation of various products with distinct morphologies is illustrated in Scheme 1. As mentioned above, Cu(OH)2 is not stable. When heated at 70 °C for 10 min, it is transformed solely into black CuO, which has a sheet-like morphology and a submicrometer size. Upon the addition of a small amount of N2H4·H2O, an obvious change in the product morphology and size is observed (Fig. 3b, SEM image). Clearly, there should be a dissolution–recrystallization process starting from the solid precursor.26 During the reduction process, the added N2H4·H2O reacts with the free Cu2+ ions via coordination and reduction steps. With the progressive dissolution of CuO to yield Cu2+ and the subsequent reduction of Cu2+ to Cu+, the CuO particles are gradually dissolved and recrystallization occurs.25,26 When the amount of N2H4·H2O reaches 1.5 mL, part of the CuO is transformed into Cu2O, forming nanocrystals of both CuO and Cu2O with premature polyhedron structures. Continuously increasing the amount of N2H4·H2O to 2.5 mL, CuO is completely transformed into Cu2O, and even part of the yielded Cu2O is further converted into Cu. Thus, the coexistence of CuO, Cu2O, and Cu grains with structures of spheres, cubes, octahedra and other polyhedra is obtained. It should be noted that the presence of small amounts of ions such as Na+, OH−, etc. has a great influence on the morphology of the Cu2O product.26 Finally, with a further increase of the N2H4·H2O amount to 3.5 mL or 5.0 mL, because there is a surplus of the reducing agent, the generated Cu2O is converted into Cu2O–Cu or pure Cu completely. Therefore, by tuning the supply of N2H4·H2O, it is possible to control the content of CuO, Cu2O, and Cu in the final Cu-based products, as proved by the above comprehensive characterizations with XRD, TG, TPR, XPS and TEM techniques.
 |
| | Scheme 1 Graphic description of the reduction mechanism. | |
3.3. Catalytic property of Cu-based catalysts
Table 2 summarizes the catalytic performance of all the samples for M2 synthesis via the Rochow reaction. As shown, the CuO sample achieves a M2 selectivity of 66.1% and a Si conversion of 26.4%. For CuO–Cu2O, the corresponding values become 68.7 and 27.2%, respectively. In the case of CuO–Cu2O–Cu, a markedly improved M2 selectivity of 80.0% and Si conversion of 51.4% are achieved, higher than those of Cu2O–Cu (71.8% M2 selectivity and 32.0% Si conversion). For sole Cu, both the M2 selectivity and the Si conversion are obviously decreased again. Also, the characterization data of the reaction product organosilanes via GC are provided (see Tables S1–S5 and Fig. S1–S5 in ESI†). It should be pointed out that the error bars for both conversion and selectivity were ±0.1% and all the catalytic data were obtained by at least three repeated experiments using the same silicon. As we know, M2 is the most desirable monomer in the organosilane industry, and the above results demonstrate that the prepared catalysts possessing the three components of Cu, Cu2O, and CuO have the best M2 selectivity and yield among all the samples, probably because of the synergistic effect between Cu, Cu2O, and CuO, which promotes the catalytic activity for the Rochow reaction.5 In addition, byproducts such as M1, M3, M1H, M2H, LB, and HB with a negligible difference over the different prepared catalysts are also detected, except for the Cu sample.
Table 2 Catalytic performances of all the catalysts for the Rochow reactiona
| Samples |
Product composition (%) |
| M1 |
M2 |
M3 |
M1H |
M2H |
LBR |
HBR |
M2 yield |
C–Si (%) |
| Reaction conditions: cat., 1.0 g; catalyst: Si (mass ratio) = 1:10; flow rate of CH3Cl, 25 mL min−1; temp., 325 °C; time, 24 h. |
| CuO |
12.5 |
66.1 |
1.4 |
14.9 |
0.8 |
0.3 |
4.0 |
17.5 |
26.4 |
| CuO–Cu2O |
11.3 |
68.7 |
1.0 |
9.2 |
0.4 |
0.4 |
9.0 |
18.7 |
27.2 |
| CuO–Cu2O–Cu |
10.1 |
80.0 |
1.5 |
7.2 |
0.9 |
0.1 |
0.2 |
41.1 |
51.4 |
| Cu2O–Cu |
11.4 |
71.8 |
1.0 |
11.2 |
1.1 |
0.1 |
3.4 |
23.0 |
32.0 |
| Cu |
21.9 |
47.3 |
1.0 |
23.0 |
0.9 |
0.3 |
5.6 |
8.8 |
18.7 |
Fig. 5 displays the XRD patterns of the waste contact masses after the reaction, which contain the unreacted Si, the Cu-based catalyst, and a trace amount of the promoter Zn. As shown in Fig. 5a, all the waste contact masses are composed of Si and Cu, but lack the Cu2O and CuO species. The formation of Cu may originate from the reaction of MeCl with the lattice oxygen of the Cu-based catalyst.9 An enlarged view of the XRD patterns in the range of 40–50° (Fig. 5b) shows the presence of η-Cu3Si and Cu6.69Si species, suggesting the formation of alloyed CuxSi active components by the reaction of Cu and Si via diffusion during the reaction. The Cu3Si alloy, generally produced between the Cu catalyst and the Si interface at elevated temperatures,29 is normally regarded as the key catalytic active species in the Rochow reaction,30,31 by which methylchlorosilanes (MCSs), especially M2, are formed. The amount of Cu3Si can substantially affect the Si conversion and M2 selectivity.6 The intensities of the Cu3Si peaks observed for CuO–Cu2O–Cu are much higher than that of the other samples, suggesting that CuO–Cu2O–Cu is more active in generating Cu3Si than the other samples. This may be because the ternary composition of CuO–Cu2O–Cu has a stronger synergistic effect and closer contact between the catalysts and the solid Si, thus promoting the formation of more active Cu3Si phases, which is the key step to enhance the catalytic activity towards M2 production.
 |
| | Fig. 5 XRD patterns of the waste contact masses (a) and enlarged view in the 2θ range of 40–50° (b). | |
Fig. 6a shows an SEM image of the Si particles after the reaction using CuO–Cu2O–Cu as the catalyst. The smooth region on the surface of the Si particles stems from the unreacted Si, while the coarse or cavity-like region comes from reacted Si, suggesting the occurrence of etching process during the reaction, which is consistent with the so-called anisotropic etching reaction mechanism.14 The element mapping images show the distribution of the elements Si (Fig. 6b), Cu (Fig. 6c) and C (Fig. 6d) on the surface of the waste contact mass. The element Si mapping image clearly shows the reacted (dark yellow) and unreacted (bright yellow) zones of the Si particles, while both Cu (dark cyan) and C (red) are distributed uniformly on the reacted or etched Si surface, indicating the occurrence of a catalytic reaction between the Si particles and Cu-based catalysts. As shown in Scheme 2, a mechanism for the catalytic cycle is proposed. The ternary Cu–Cu2O–CuO catalyst is first transformed to Cu* (active species) via the reaction of MeCl with the lattice oxygen, which then reacts with Si to form the alloyed CuxSi active intermediate. Then, MeCl is adsorbed on the surface and transformed into M2, and meanwhile Cu* is released again.
 |
| | Fig. 6 SEM image of Si particles after reaction (a), elemental mapping images of Si (b), Cu (c) and C (d). | |
 |
| | Scheme 2 A proposed catalytic mechanism for the catalytic cycle. | |
The above results allow us to describe graphically the possible Rochow reaction process in the presence of a Cu-based catalyst (Scheme 3). First, Si is mixed with the reduced Cu particles in submicrometer size before the reaction. With diffusion of Cu into the Si, the CuxSi alloy is gradually formed at the Cu catalyst and the Si interface at elevated temperatures; the alloy then further reacts with the gas MeCl to form MSCs. As the reaction proceeds further, more elemental copper is produced while the Cu in the CuxSi alloy is reduced until deactivation of the catalyst occurs. At the same time, MeCl may be transformed into carbon depositions (C), initiated by the metallic Cu, leading to the deactivation of the contact mass. The presence of the multicomponent particle structure should enhance the gas diffusion into the Cu–Si contact area, resulting in the formation of much more of the active species. Nevertheless, the real synergistic effect is not yet fully understood at present and should be further explored.
 |
| | Scheme 3 Schematic illustration of the process of the Rochow reaction. | |
This method is simple without use of any complicated and delicate equipment, and the reaction conditions are mild, as the reaction temperature is only 70 °C. More importantly, this method is readily scalable. Our experiment results showed that about 10 g of products could be obtained at one time by using a 500 mL beaker (not shown), indicating its great potentiality for industrial application.
4. Conclusions
In summary, we have demonstrated the controllable synthesis of Cu-based catalysts with variable CuO, Cu2O and Cu content by a wet chemical method, in which the key is to control the amount of N2H4·H2O, which is used as the reducing agent. Various characterizations indicate that the reduction of CuO is a successive reaction that yields Cu2O and Cu, which in turn provides the possibility to finely tune the catalyst composition via varying the amount of reducing agent. Comparing with the synthesized CuO and Cu particles, the multicomponent CuO–Cu2O–Cu catalyst shows much higher M2 selectivity and Si conversion in the Rochow reaction, proving the significance of the synergistic effect among the components. It is expected that these ternary CuO–Cu2O–Cu catalysts will have great potential in the industrial Rochow reaction.
Acknowledgements
The authors gratefully acknowledge the financial supports from the National Natural Science Foundation of China (no. 51272252 and 21206172). Z. Z. is grateful to the kind supports of both Nanyang Technological University (NTU) and Institute of Chemical Engineering and Sciences (ICES) under Agency for Science, Technology and Research (A*STAR).
References
- E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963–965 CrossRef CAS.
- A. D. Gordon, B. Hinch and D. R. Strongin, Catal. Lett., 2009, 133, 14–22 CrossRef CAS.
- H. Lieske and R. Zimmermann, Catal. Lett., 1995, 33, 413–420 CrossRef CAS.
- L. Zhang, S. Hao, C. H. Yang, J. Li, K. Yang, C. F. Hu and S. B. Ge, Appl. Organomet. Chem., 2011, 25, 508–513 CrossRef CAS PubMed.
- Z. L. Zhang, H. W. Che, Y. L. Wang, J. J. Gao, L. R. Zhao, X. L. She, J. Sun, P. Gunawan, Z. Y. Zhong and F. B. Su, Ind. Eng. Chem., 2012, 51, 1264–1274 CrossRef CAS.
- L. N. Lewis and W. J. Ward, Ind. Eng. Chem., 2002, 41, 397–402 CrossRef CAS.
- D. H. Sun, B. E. Bent, A. P. Wright and B. M. Naasz, J. Mol. Catal. A: Chem., 1998, 11, 169–183 CrossRef.
- D. H. Hashiguchi, R. J. Dietrich and G. P. Schoepe, US Pat., 4504596, 1985.
- L. N. Lewis, W. V. Ligon and J. C. Carnahan, Silicon Chem., 2002, 1, 23–33 CrossRef CAS.
- A. D. Gordon, B. Hinch and D. R. Strongin, J. Catal., 2009, 266, 291–298 CrossRef CAS.
- K. H. Brookes, M. R. H. Siddiqui, H. M. Rong, R. W. Joyner and G. J. Hutchings, Appl. Catal., A, 2001, 206, 257–265 CrossRef CAS.
- Y. X. Zhu, Y. L. Wang, L. Y. Song, X. Chen, W. Y. Liu, J. Sun, X. L. She, Z. Y. Zhong and F. B. Su, RSC Adv., 2013, 3, 9794–9802 RSC.
- Z. L. Zhang, H. W. Che, Y. L. Wang, X. L. She, J. Sun, P. Gunawan, Z. Y. Zhong and F. B. Su, ACS Appl. Mater. Interfaces, 2012, 4, 1295–1302 CAS.
- Z. L. Zhang, H. W. Che, J. J. Gao, Y. L. Wang, X. L. She, J. Sun, P. Gunawan, Z. Y. Zhong and F. B. Su, Catal. Sci. Technol., 2012, 2, 1207–1212 CAS.
- Z. L. Zhang, H. W. Che, Y. L. Wang, L. Y. Song, Z. Y. Zhong and F. B. Su, Catal. Sci. Technol., 2012, 2, 1953–1960 CAS.
- Z. L. Zhang, H. W. Che, J. J. Gao, X. L. She, J. Sun, Z. Y. Zhong and F. B. Su, RSC Adv., 2012, 2, 2254–2256 RSC.
- X. Chen, L. H. Jia, Y. L. Wang, L. Y. Song, Y. X. Zhu, W. Y. Liu, Z. Y. Zhong and F. B. Su, J. Colloid Interface Sci., 2013, 404, 16–23 CrossRef CAS PubMed.
- M. Khitouni, R. Daly, M. Mhadhbi and A. Kolsi, J. Alloys Compd., 2009, 475, 581–586 CrossRef CAS PubMed.
- W. Y. Liu, L. H. Jia, Y. L. Wang, L. Y. Song, Y. X. Zhu, X. Chen, Z. Y. Zhong and F. B. Su, Ind. Eng. Chem., 2013, 52, 6662–6668 CrossRef CAS.
- S. M. Liu, Y. L. Wang, Y. X. Zhu, G. N. Wang, Z. L. Zhang, H. W. Che, L. H. Jia and F. B. Su, RSC Adv., 2014, 4, 7826–7833 RSC.
- F. Capece, V. D. Castro, C. Furlani, G. Mattogno, C. Fragale, M. Gargano and M. Rossi, J. Electron Spectrosc. Relat. Phenom., 1982, 27, 119–128 CrossRef CAS.
- H. Tolentino, F. Baudelet, A. Fontaine, T. Gourieux, G. Krill, J. Henry and J. Rossat-Mignod, Phys. C, 1992, 192, 115–130 CrossRef CAS.
- K. J. Ziegler, R. C. Doty, K. P. Johnston and B. A. Korgel, J. Am. Chem. Soc., 2001, 123, 7797–7803 CrossRef CAS PubMed.
- W. Z. Wang, O. K. Varghese, C. M. Ruan, M. Paulose and C. A. Grimes, J. Mater. Res., 2003, 18, 2756–2759 CrossRef CAS.
- A. Muramatsu and T. Sugimoto, J. Colloid Interface Sci., 1997, 189, 167–173 CrossRef CAS.
- J. W. Zhu, H. P. Bi, Y. P. Wang, X. Wang, X. J. Yang and L. D. Lu, Mater. Lett., 2008, 62, 2081–2083 CrossRef PubMed.
- J. P. Li, F. Q. Sun, K. Y. Gu, T. X. Wu, W. Zhai, W. S. Li and S. F. Huang, Appl. Catal., 2011, 406, 51–58 CrossRef CAS PubMed.
- Y. Cudennec and A. Lecerf, Solid State Sci., 2003, 5, 1471–1474 CrossRef CAS PubMed.
- N. Floquet, S. Yilmaz and J. L. Falconer, J. Catal., 1994, 148, 348–368 CrossRef CAS.
- E. Gaffet and F. Bernard, Ann. Chim. Sci. Mater., 2002, 27, 47–59 CrossRef CAS.
- D. H. Sun, B. E. Bent, A. P. Wright and B. M. Naasz, Catal. Lett., 1997, 46, 127–132 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10999j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.