
Synthesis and properties of novel diisopropyl-functionalized polyglycolide–PEG copolymers†
Abstract
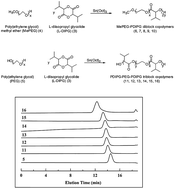
Because of the importance of glycolide-based polymers in materials and medical applications, we have synthesized alternative novel thermosensitive biomaterials to be possible candidates instead of the well-known polymers. The synthesis of L-diisopropyl glycolide monomer was performed in two steps, novel PEG based poly(diisopropyl glycolide) diblock and triblock copolymers (MePEG–PDIPG, PDIPG–PEG–PDIPG) were synthesized with high conversions by ring opening polymerization. The molar mass distributions of the copolymers were very narrow, and the NMR measurements confirmed that no racemization had occurred in the polymer synthesis even at 180 °C. Gel–sol experiments were conducted after each component’s length in MePEG–PDIPG and PDIPG–PEG–PDIPG was adjusted with care during the synthesis. The polymer showed a sol property at 42–45 °C, which is suitable for injection and a gel property at body temperature. After loading paclitaxel into gels effectively, the anticancer drug showed prolonged release (60 days). SEM analysis confirmed that the gels turned into a more porous structure due to drug release.
 Please wait while we load your content...
Please wait while we load your content...

