
Uncapped Au–Pd colloidal nanoparticles show catalytic enhancement
Abstract
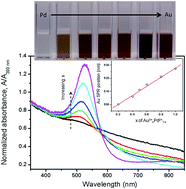
The catalytic properties of Pd have triggered a strong interest in the related catalysis by Au–Pd nanoparticles. However, the analysis of such phenomena has been complicated so far due to the presence of capping. Using X-ray irradiation, we produced uncapped Au–Pd nanoparticles and studied their catalytic features, finding in particular their relationship to the Pd content. Furthermore, the fabrication process is per se interesting, yielding excellent and flexibly controllable nanoparticles with a rather simple procedure.
 Please wait while we load your content...
Please wait while we load your content...

