
Ni nanoparticles supported on mesoporous silica (2D, 3D) architectures: highly efficient catalysts for the hydrocyclization of biomass-derived levulinic acid†
Mohan Varkolu‡
*,
Venkateshwarlu Velpula,
Saidulu Ganji,
David Raju Burri and
Seetha Rama Rao Kamaraju*
Inorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Hyderabad, India-5000071. E-mail: ksramarao@iict.res.in; Fax: +91-40-27160921; Tel: +91-40-27191712
First published on 24th June 2015
Abstract
Ni nanoparticles supported on various mesoporous silicas with 2D (COK-12) and 3D architectures (KIT-6 and SBA-16) exhibited superior catalytic performance in the vapor-phase hydrocyclization of biomass-derived levulinic acid at atmospheric pressure. The catalysts were systematically characterized by XRD, N2 physisorption, FTIR, SEM-EDAX, NH3-TPD, TPR, H2 chemisorption, HRTEM, and XPS techniques. The retention of the mesoporous structure of the catalysts was confirmed by the low angle XRD, pore size distribution, SEM, and TEM. TPR results suggested the presence of Ni particles inside the 3D-pore architecture of SBA-16 and KIT-6. Whereas in the case of 2D-architectured silica (COK-12), most of the Ni particles were laid on the surface of the support and/or no particles were found in the inside of the pore. The physico-chemical characteristics like confinement-effect and interconnected network of the catalysts play a crucial role in the catalytic activity and product distribution. Deep hydrogenation of levulinic acid to 2-methyl tetrahydrofuran was observed over Ni supported on 3D architectured silicas (KIT-6 and SBA-16) which can be attributed to confinement of Ni nanoparticles in 3D architectured silicas.
Introduction
The guaranteed continuous availability of biomass concentrates our attention on the transformation of biomass/biomass-derived platform molecules to useful chemicals/fuels. Recent statistics revealed the lifetime of non-renewable sources such as crude oil, natural gas, and coal were 46, 58 and 116 years, respectively.1 In the scenario of a future fuel crisis due to the depletion of fossil fuel resources, it is necessary to search for alternative fuels. Utilization of biomass is one of the options as of now, because of its availability at an affordable price and carbon neutrality maintenance. The biomass-derived platform molecules, levulinic acid (LA), hydroxyl methyl furfural (HMF), cellulose, glucose, and fructose are just to name a few. Among these platform molecules, LA occupies the top 12 potential molecules2,3 because of its simple accessibility through the acid hydrolysis of cellulosic biomass4–8. The presence of highly reactive groups such as –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH, makes attractive the transformation of levulinic acid to a large number of fuels, fuel additives and chemicals.9 A plethora of research has been devoted to the conversion of levulinic acid to various products viz, γ-valerolactone, 2-methyl tetrahydrofuran, alkyl levulinates, N-alkyl pyrrolidine, 1,4-pentanediol, diphenolic acid, acrylic acid and acetyl acrylic acid etc. Among these products, γ-valerolactone is regarded as an important chemical due to its variety of uses such as fuel additive, solvent, insecticide, adhesive, cutting oil, brake fluid, coupling agent in dyes and its conversion to yield diesel fuel, jet fuel and other fuels. γ-Valerolactone can be prepared by the hydrogenation of levulinic acid in the presence of homogeneous and heterogeneous catalysts.10,11 Although homogeneous catalysts are efficient, they have certain disadvantages like separation of the catalyst after the reaction and recyclability. To overcome this problem, various heterogeneous catalysts have been used in the levulinic acid hydrogenation in a batch mode.10 However, continuous process is more favorable in view of green chemistry requirements like removal of toxic and volatile organic solvents.
O and –COOH, makes attractive the transformation of levulinic acid to a large number of fuels, fuel additives and chemicals.9 A plethora of research has been devoted to the conversion of levulinic acid to various products viz, γ-valerolactone, 2-methyl tetrahydrofuran, alkyl levulinates, N-alkyl pyrrolidine, 1,4-pentanediol, diphenolic acid, acrylic acid and acetyl acrylic acid etc. Among these products, γ-valerolactone is regarded as an important chemical due to its variety of uses such as fuel additive, solvent, insecticide, adhesive, cutting oil, brake fluid, coupling agent in dyes and its conversion to yield diesel fuel, jet fuel and other fuels. γ-Valerolactone can be prepared by the hydrogenation of levulinic acid in the presence of homogeneous and heterogeneous catalysts.10,11 Although homogeneous catalysts are efficient, they have certain disadvantages like separation of the catalyst after the reaction and recyclability. To overcome this problem, various heterogeneous catalysts have been used in the levulinic acid hydrogenation in a batch mode.10 However, continuous process is more favorable in view of green chemistry requirements like removal of toxic and volatile organic solvents.
Nowadays, mesoporous materials found to be attract considerable attention owing to their potential applications in the adsorption, separation, and catalysis etc., due to their high surface area, high porosity, uniform pore size and diffusion limitations. These features provide a consistent and well-isolated environment for the growth of nanoparticles, which is of particular interest for size-dependent reactions.12–18
In continuation of our ongoing research in the development of Ni-based catalysts for hydrocyclization of levulinic acid in continuous process at atmospheric pressure.19–22 The present work aims the application of nickel nanoparticles supported on various 2D and 3D-architectured mesoporous silicas for the vapor phase hydrocyclization of levulinic acid. Elucidation of confinement of Ni particle and architecture of silica on the catalytic activity and product distribution is also the one of the objectives of the present study.
Experimental
Preparation of KIT-6
KIT-6 mesoporous silica was prepared according to the procedure described elsewhere.23 In a typical synthesis, a homogeneous solution was obtained by dissolving 32.0 g of P123 (M/s. Sigma-Aldrich Chemicals, USA) and 63.0 g of 35.0 wt% hydrochloric acid in 1150 g of deionized water under magnetic stirring for 8 h at 35 °C. To this solution, 32 g of 1-butanol was added followed by stirring for 1 h at 35 °C, 68.8 g of tetraethylorthosilicate (TEOS) was added at once to the P123-butanol–HCl solution. This mixture was continuously stirred at 35 °C for 24 h, for the formation of KIT-6 silica phase. The mixture containing KIT-6 was then heated at 130 °C for 24 h under static conditions in a Teflon-lined autoclave. The temperature aging process was applied to broaden the silica pore diameters. The KIT-6 product was filtered and dried at 100 °C in air. The product was washed with an HCl–ethanol mixture to extract the P123 as much as possible and was subsequently calcined at 550 °C for 5 h in air.Preparation of COK-12
The ordered mesoporous silica, i.e., COK-12 was prepared by self-assembly method24 using long chain ionic surfactant, i.e., P123 as template and sodium silicate as SiO2 source which was subsequently used as support for the Ni catalyst. In a typical procedure, COK-12 was prepared by dissolving the 4 g of the triblock copolymer pluronic P123 (M/s. Sigma Aldrich Chemicals, USA) in 107.5 ml of water. To this solution 3.684 g of citric acid monohydrate (M/s. S. D. Fine chemicals, India) and 2.54 g trisodium citrate (M/s. S. D. Fine chemicals, India) were added. The resulting surfactant solution was stirred for 24 h. 10.4 g sodium silicate solution (10% NaOH, 27% SiO2 M/s. Merck Germany) was diluted with 30 g of H2O and added to the surfactant solution. The pH was measured prior to and after sodium silicate addition. The solution was stirred for 5 min at 175 rpm with a mechanical stirrer and kept at room temperature (20 °C) without agitation for 24 h. The synthesized material was filtered with several washings using deionized water and dried at 60 °C overnight in an air. Finally, the support was calcined in the air in two steps, 8 h at 300 °C and 8 h at 500 °C with 1 °C min−1 ramping.Preparation of SBA-16
SBA-16 was prepared by using a triblock copolymer surfactant (EO106PO70EO106, called F127 (M/s. Sigma Aldrich chemicals, USA)) as a structure directing agent, tetraethylorthosilicate (TEOS, M/s. Sigma-Aldrich chemicals, USA) as silica source at low acid concentration without using NaCl salt.25 In a typical procedure, 3.72 g of F127 was dissolved in 139 ml 0.5 M HCl at 40 °C. After 2–3 h stirring, 18 ml of TEOS was added dropwise. The solution containing molar composition = 1 TEOS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.00367 F127:0.864 HCl:100.231H2O was stirred for 20 h at 40 °C, and subsequently transferred to Teflon bottle and aged at autogenous pressure for 24 h at 100 °C. The material was filtered with several washings with deionized water and dried at 80 °C, and then calcined at 550 °C for 4 h with a heating rate of 3 °C min−1.
:0.00367 F127:0.864 HCl:100.231H2O was stirred for 20 h at 40 °C, and subsequently transferred to Teflon bottle and aged at autogenous pressure for 24 h at 100 °C. The material was filtered with several washings with deionized water and dried at 80 °C, and then calcined at 550 °C for 4 h with a heating rate of 3 °C min−1.
Preparation of mesoporous silica supported nickel catalysts
Mesoporous silica (KIT-6, COK-12 and SBA-16) supported Ni catalysts with 30 wt% Ni loadings were prepared by wet impregnation method. In a typical synthesis procedure, the 3.5 g of mesoporous silica and 7.4 g of Ni(NO3)2·6H2O (AR grade obtained from M/s. Loba Chemie, India) was dissolved in deionized water and kept it on hot plate at 80 °C, stirred at a rate of 300 rpm until the complete dryness. The prepared catalysts were dried overnight at 100 °C and calcined at 450 °C for 5 h in an air. The prepared catalysts Ni/KIT-6, Ni/COK-12, and Ni/SBA-16 were designated as NK6, NC12 and NS16 respectively. The mesoporous silica supports KIT-6, COK-12, and SBA-16 were designated as K6, C12, and S16 hereafter respectively.Characterization of the catalysts
Powder X-ray diffraction (XRD) patterns of all the catalysts were recorded on a Ultima IV diffractometer (M/s. Rigaku Corporation, Japan) with a scanning rate of 0.02° using Ni filtered Cu Kα radiation (λ = 1.5406 Å) with a scan speed of 4° min−1 and a scan range of 10–80° at 40 kV and 20 mA.The BET surface area, pore size, and pore volume of all the catalysts were performed on a Quantachrome Quadrasorb SI by N2 adsorption–desorption at liquid N2 temperature.
The morphological studies of catalysts were carried by both S-4800 scanning electron microscope (M/s Hitachi Company, Japan) along with energy dispersive X-ray analysis (EDS) (LINK ISIS-300, M/s. Oxford) with an accelerating voltage of 15 kV.
The TEM images were obtained on a JEOL JEM 2000EXII apparatus operating between 160 and 180 kV respectively. For TEM analysis, the samples were suspended ultrasonically in ethanol and then dropped onto the carbon coated copper grid, the solvent was then evaporated in an air oven at 80 °C for 6 hours.
Temperature programmed reduction (TPR) was carried out on a homemade system to evaluate reducing behavior of the nickel and also metal support interaction or location of the metallic particle either in outside or inside of the pores of support. Prior to TPR analysis, the catalyst was heated to 140 °C at 20 °C min−1 and held for 30 min to remove the moisture, then bring down to 40 °C followed by heating to 900 °C at 20 °C min−1 in a flow of gas consisting of 5% H2 balance Argon (50 ml min−1).
X-ray photoelectron spectroscopy (XPS) data were obtained using AXIS 165 apparatus (M/s. Kratos Instruments, UK) with Al Kα target and 1486.6 eV energy at a maximum power of 12.5 kV and 16 mA current. The surface of the sample was cleaned using a 5 kV argon ion gun.
The FTIR spectra was recorded on a spectrum GX spectrometer (M/s. Perkin-Elmer, Germany) in the scan range of 4000–400 cm−1. Furthermore, pyridine adsorbed FTIR spectra was obtained for which procedure was described in our previous publication.19
Temperature programmed desorption of ammonia of the catalysts were measured by means of AUTOSORB-iQ, automated gas sorption analyser (M/s. Quantachrome Instruments, USA) to know the acidity. Prior to desorption, the catalyst samples has been soaked with the ammonia (5% NH3 balance helium) at 80 °C for 30 minutes, followed by removal of the physisorbed NH3 through the stripping with helium at the same temperature for 30 minutes. Afterwards, the temperature was raised to 800 °C at 10 °C min−1, with simultaneously monitoring of the desorbed NH3 with the inbuilt TCD in a flow of helium (60 ml min−1). The number of surface Ni sites was obtained by H2 chemisorption, measured on an AUTOSORB-iQ, an automated gas sorption pulse analyzer (M/s. Quantachrome Instruments, USA). Prior to chemisorption, the catalyst was reduced in situ under pure H2 flow by heating up to 500 °C with a heating rate of 10 °C min−1, held for 2 h, followed by evacuation for 2 h, and cooling to 40 °C. Multiple pulses of pure hydrogen were injected onto the catalyst through a septum with 100 μL syringe until saturation was observed.
Activity test
Catalytic tests were carried out in a fixed-bed quartz reactor (14 mm id and 300 mm long) at atmospheric pressure. In each catalytic run, 1 g of the catalyst and same amount of the quartz particles were loaded at the centre of the quartz reactor. Prior to the reaction, the catalysts was reduced at 500 °C for 4 h in a H2 flow of 30 ml min−1. Then the temperature was brought down to the reaction temperature where levulinic acid was fed at a flow rate of 1 ml h−1 using a syringe feed pump (M/s. B. Braun, Germany) with a WHSV = 1 h−1 along with H2. The product mixture was collected in an ice cooled trap at the regular intervals and analyzed by FID equipped gas chromatograph, GC-17A (M/s. Shimadzu Instruments, Japan) and confirmed by GC-MS, QP-5050 (M/s. Shimadzu Instruments, Japan).Results and discussion
X-ray diffraction (XRD) studies
Fig. 1 depicts the low angle XRD patterns of mesoporous silica supported Ni catalysts calcined at 450 °C for 5 h which clearly demonstrates the conservation of its structural integrity even after deposition of Ni (30 wt%). It is significant that the conservation of structural reliability during the entire process (impregnation, drying, and calcination) to get final catalyst is an important feature.13,14,26 The NC12 catalyst showed well-resolved diffraction patterns which were indexed as (100), (101) and (200) planes of hexagonal space group symmetry p6mm.27,28 The low angle XRD patterns of NS16 catalyst demonstrates a prominent peak at 0.89° (110) and other two small peaks at 1.25° (200) and 1.54° (211) on a 2θ scale and signifying the presence of ordered mesopores with body-centered cubic symmetry (Im3m).28,29 While the NK6 catalyst demonstrates a reflections, in the low angle region, which could be assigned to the (211) and (220) planes of 3D cubic structure indicative of retaining its ordered mesopores ascribed to Ia3d symmetry even after Ni deposition.16,18 | ||
| Fig. 1 Small angle XRD patterns of calcined mesoporous silica supported Ni catalysts. | ||
The XRD patterns of all the calcined catalysts (Fig. 2) indicates the presence of NiO crystalline phase at 37.29°, 43.4°, 62.87°, 75.41° and 79.40° only on 2-theta scale. The XRD patterns of the three reduced catalysts are shown in Fig. 3 and the results suggest that the metallic Ni crystallites exist in FCC structure with the following planes: 44.5°, 53.0° and 78.3°. It is well reported in the literature12–18,27–31 that the preservation of the mesoporous structure of silica by doping of various metals, even with higher amounts. The present results are in good agreement with the earlier reports. The broad peak noticed in both calcined and reduced catalysts at a 2θ value of ∼22° corresponds to amorphous silica.32
 | ||
| Fig. 2 Wide angle XRD patterns of calcined mesoporous silica supported Ni catalysts. ICDD. no. 78-0429 for NiO. | ||
 | ||
| Fig. 3 Wide angle XRD patterns of reduced mesoporous silica supported Ni catalysts. ICDD. no. 88-2326 for Ni. | ||
N2 adsorption–desorption isotherms of the catalysts are shown in Fig. 4. The results (Fig. 4) indicate a type IV isotherm with H1 hysteresis loop, which is a characteristic of mesoporous material with uniform porosity defined by IUPAC. A sharp inflection in P/P0 range from 0.5 to 0.8 is found in the isotherms of all three mesoporous silica supports as well as Ni incorporated mesoporous silica catalysts providing further evidence of maintaining their mesoporous nature even after incorporation of Ni.33–35 The pore size distribution pattern is shown in Fig. 5. The uni-model pore distribution in a diameter range of 5–10 nm is observed in K-6 sample while in case of NK6 a bi-model pore distribution is observed after the incorporation of Ni. This could be possible whenever the Ni is present in the inside of the pores. In addition, we found the decrease in pore volume in NK6 catalyst which also suggests the presence of Ni particles in the pores. Similarly, a broad range of (4–8 nm) pore diameter is observed in C12 and NC12. Furthermore, the consistency in pore volume in case of C12 and NC12 implies the presence of metal particles on the surface of C12 support. Accumulation of pores in the pore diameter range of 4–5 nm in S16 and NS16 catalysts clearly indicates the presence of uniform mesoporous structure. By the incorporation of nickel, an apparent decrease in surface area and an enlargement in pore diameter was observed in NS16 catalyst. This enlargement in pore diameter can be explained by the incorporation of nickel into silica framework, which leads to the considerable contraction of walls. As a result, an enlargement in pore diameter were observed.36 Likewise, we also observed the lower pore wall thickness of NS16 than the S16.
 | ||
| Fig. 4 N2 adsorption–desorption isotherms of mesoporous silicas and Ni incorporated mesoporous silicas. | ||
 | ||
| Fig. 5 Pore size distribution of mesoporous silicas and Ni incorporated mesoporous silicas. | ||
Decrease in BET surface area was observed upon the impregnation of Ni (Table 1) onto the respective supports which reveal that most of the active component is accumulated either inside the pores or blocked the pores. The preservation of the long-range order of mesoporous silicas (K6, C12, and S16) upon impregnation of Ni (30 wt%) is evident from pore size distribution curves. These results are in line with the results of low angle XRD. The pore size of K6 has been decreased by the incorporation of Ni, which in turn reflected in an increase in the pore wall thickness. The pore wall thickness of all these mesoporous materials are in the range of 2–8 nm which is in good agreement with the previous reports.34,35 It was reported that the deposition of Ni (10 wt%) on SBA-15 support by liquid phase reductive deposition did not alter the mesoporous structure of SBA-15.12 An analogous scrutiny was made in the case of Ru/SBA-15 catalysts.31 Intactness of SBA-15 structure was reported in Ni/SBA-15 catalysts.34
| Catalyst | SBET (m2 g−1) | Vp (cc g−1) | Dp (nm) | d(100)a nm | a0b nm | Pore wall thickness (t = a0 − Dp) nm |
|---|---|---|---|---|---|---|
| a Obtained from the (100) plane in the low angle XRD patterns.b a0 = 2d100 √3, SBET = BET surface area, Dp = BJH pore size. | ||||||
| K6 | 651.1 | 0.88 | 7.4 | 8.8 | 10.2 | 2.8 |
| C12 | 297.4 | 0.36 | 4.6 | 8.2 | 9.4 | 4.8 |
| S16 | 387.7 | 0.31 | 4.0 | 10.0 | 11.6 | 7.6 |
| NK6 | 301.2 | 0.42 | 4.6 | 8.2 | 9.4 | 4.9 |
| NC12 | 187.4 | 0.25 | 4.5 | 9.0 | 10.4 | 5.9 |
| NS16 | 319.2 | 0.26 | 4.5 | 10.3 | 11.9 | 7.3 |
In order to investigate the metal support interactions, location of the metal particles (inside the pores or on the surface) and reduction behavior of the catalyst, temperature programmed reduction (TPR) is performed and the results are depicted in Fig. 6. The catalysts NK6 and NS16 show two predominant reduction maxima, one at low temperature, and the other at high temperature. Whereas NC12 show only single reduction peak lower temperature. The lower temperature peak observed at around 420 °C can be attributed to the reduction of bulk NiO,37–40 whereas the high temperature peak noticed at around 650 °C can be ascribed to the reduction of smaller NiO particles located within the pores.41,42 Furthermore, the hydrogen consumption was calculated by integration of the area under the TPR peaks. The hydrogen consumption values at lower temperature were 227 and 345 μmol g−1 for NK6 and NS16, while the hydrogen consumption at higher temperature were 74 and 126 μmol g−1 for NK6 and NS16 respectively. On the other hand, 612 μmol g−1 hydrogen is consumed by NC12. Wu et al., reported that the NiO particles located inside the pores have strong interaction with MCM-41 than the bulk NiO particles and found to be reduced at higher temperatures.41 In addition, they also explained the discrepancy in location of NiO particles, through the preparation of two types of Ni/MCM-41 catalysts, one with NiO particles located inside the pores and the other with NiO particles located outside the pores and confirmed by the temperature difference in their TPR profiles.42 The TPR results in the present case, clearly demonstrates the presence of NiO particles in both inside and outside the pore network particularly in the 3-dimentional pore network catalysts; whereas the NiO particles are only appeared on the surface or outside of the pore network in 2-dimentional pore network catalysts. The present results are in good agreement with the results of Wu et al.41,42 Besides, Hodgkins et al. found maghemite nanocrystals inside the mesopores even they used impregnation method as a synthesis procedure.43 So, one can understand that the impregnation method itself can enable to provide the particles in the inside of the pores when porous materials used as a support. Furthermore, it is well known that there is a high possibility to diffuse the smaller metal particles through the 3D architectures rather than the 2D architectures. In fact, one can understand from the dispersion and particle size (as evidenced from the H2 chemisorption), that the average particle size found in the NK6 and NS16 is lower compared to NC12 catalyst. The presence of Ni particles in the inside of the pores in 3D architectures and surface or outside of the pores in 2D architectures could be presumably due to the variation in the particle sizes (i.e., lower than the size of the pore) and outside particles (i.e., higher than the size of the pore) although the composition is same in all the catalysts.
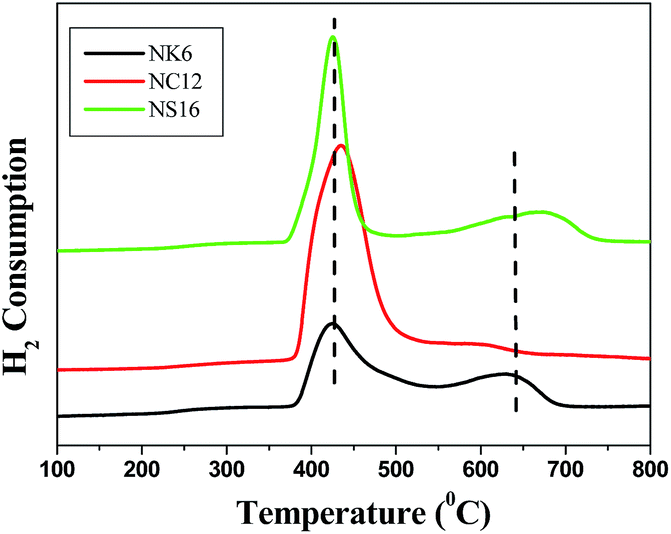 | ||
| Fig. 6 TPR profiles of Ni incorporated mesoporous silicas. | ||
H2 pulse chemisorption has been performed to assess the Ni dispersion, active metal surface area, average particle size, and the number of surface active species presented in the catalysts. The hydrogen uptake (Nm), active metal surface area (AMSA), dispersion (D) and average particle size were calculated by using TPRWin software and tabulated in Table 2. Higher Ni dispersion was observed for NK6 (14.44) than that of NS16 (12.25) and NiC12 (11.49). The average particle size of Ni in NK6 is 7 nm and NS16 is 8.25 nm are smaller than that in NC12 (8.80 nm). The high dispersion of Ni is noticed in the NK6 and NS16 catalysts are due to the presence smaller size Ni nanoparticles found in those catalysts compared to NC12 catalyst. These results also suggests that the AMSA is found to be very high in NK6 catalysts and are in the following order: NK6 > NS16 > NC12.
| Catalyst | Acidity (μmol g−1) | Nm (μmol g−1) | AMSA (m2 g−1 catalyst) | AMSA (m2 g−1 Ni) | Average particle size (nm) | D (%) |
|---|---|---|---|---|---|---|
| NK6 | 88.4 | 368.97 | 28.86 | 96.21 | 7.00 | 14.44 |
| NC12 | 61.2 | 293.55 | 22.96 | 76.54 | 8.80 | 11.49 |
| NS16 | 73.8 | 313.19 | 24.50 | 81.67 | 8.25 | 12.25 |
Fig. 7 shows TEM images of mesoporous silicas supported with 30 wt% Ni. The Fig. 7a (NK6) and Fig. 7c (NS16) shows that the Ni particles are present on the surface as well as in the inside the pore network while NC12 (Fig. 7b) catalyst exhibited that the Ni particles are in uniformly dispersed on the outside the pore network. Such a homogeneous dispersion of active species in the catalysts are highly advantageous to obtain optimal catalytic activity in various applications.44 The magnified figures clearly shows the cubic (NK6 and NS16) and hexagonal (NC12) morphologies of the catalysts which indicate the retention of the porous structure even after incorporation of high amount of Ni (30 wt%) (see ESI Fig. S6†). Similarly, the morphology of C12 and NC12 remains same even after incorporation of Ni (see ESI Fig. S2†).
 | ||
| Fig. 7 TEM images of mesoporous silicas supported Ni catalysts (a) NK6 (b) NC12 (c) NS16. | ||
Activity measurements
To completely verify the reaction pathway (Scheme 1), the role of acid sites (see ESI Fig. S3 and S4†), as well as metallic sites, have been studied previously.19,20,44 It is noteworthy to mention that the reaction proceeds via Path I with the presence of both acid and metallic functions,19 while the presence of metallic sites predominantly proceeds through Path II.20 In both the paths, the ultimate products are GVL and/or valeric acid. The formation of GVL is inevitable in Path II due to easy esterification of intermediate (4-hydroxy valeric acid).20,45,46 Interestingly in the present case, we found the appearance of 2-methyl tetrahydrofuran over NK6 and NS16 catalysts; such kind of formation is conceivable due to the presence of smaller Ni nanoparticles within the 3-dimensional pores of K6 and S16 whose presence is clearly seen in TPR and TEM. | ||
| Scheme 1 Plausible reaction pathways for the levulinic acid hydrogenation. | ||
The screening of Ni-based catalysts on different supports i.e. K6, C12, and S16 for the hydrocyclization of biomass-derived levulinic acid reveals that the C12 support is found to be the best support for the production of γ-valerolactone. The catalytic activity results suggested that the NC12 catalyst is the best among the others with the GVL yield of 98% (Fig. 8). The remarkable activity of the NC12 catalyst is attributed to the Ni nanoparticles present on the cylindrical pores of the support. The deeper hydrogenated product 2-MTHF is not observed over this catalyst. While the presence of an interconnected network of pores with some of the Ni particles within the pores (as evidenced from TPR and TEM) in both NK6 and NS16 contributes to the deep hydrogenation. Though the conversion is same (100%) over all the three catalysts, the product distribution is different which depends on the pore network of the support and smaller size of Ni nanoparticles. The formation of pentanoic acid and 2-methyl tetrahydrofuran are observed (Scheme 2) over NK6 and NS16 catalysts due to the presence of smaller Ni particles within the interconnected network (Fig. 8). Plausible reaction mechanism for the formation of deeper hydrogenated products over NK6 and NS16 are shown in Scheme 2.
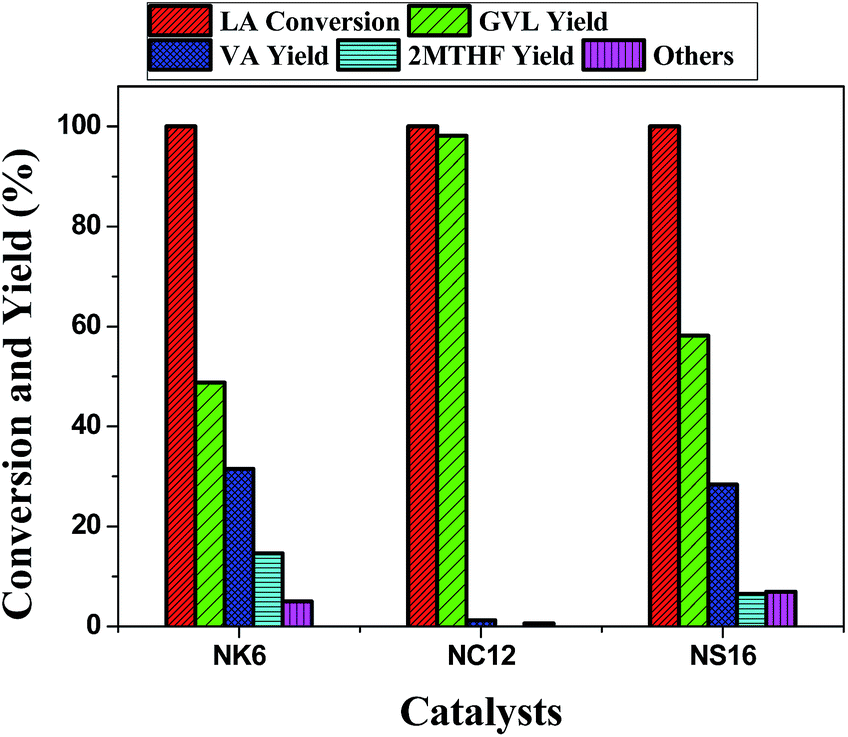 | ||
| Fig. 8 Variation in the conversion and yield against various mesoporous silica supported Ni catalysts. Reaction conditions: weight of the catalyst = 1 g, temperature = 250 °C, pressure = 1 atm, levulinic acid = 1 ml h−1, carrier gas (H2) = 1800 ml h−1, H2/LA molar ratio = 8. | ||
 | ||
| Scheme 2 Plausible reaction schemes for the hydrogenation of levulinic acid over Ni incorporated 3D mesoporous silica catalysts. | ||
Wu et al., reported that the Ni particles confined within the MCM-41 exhibited better biomass gasification activity than those present on the surface of MCM-41.42 This could be due to the easy passage of reactant molecules through the 3-dimensional porous network, thus maintaining longer residence time with Ni nanoparticles within the pores which favor the deep hydrogenation. A similar performance has been reported in an oxidation reaction over K6 and S16 supported Pd catalysts than SiO2 and SBA-15 supported Pd catalysts.18 These results suggest that the smaller Ni particles trapped within the pores of support promotes the deeper hydrogenation. Similar phenomenon can be predicted in the present case where we found the deeper hydrogenated products over NK6 and NS16 catalysts due to presence of the smaller Ni nanoparticles inside the pores which might promote the deep hydrogenation of levulinic acid. Furthermore, the Ni particles present in metallic form are evidenced from the XPS (see ESI Fig. S5†) which prevails the hydrogenation of the levulinic acid.
Effect of time on stream
To verify the stability of the catalytic systems, LA hydrogenation has been carried out for longer hours around 40 h. The catalytic performance of NK6, NC12, NS16, and Ni/SiO2 (amorphous silica supported catalyst) catalysts during the time on stream have been shown in Fig. 9. The results suggest that the there is a decrease in conversion of levulinic acid beyond 30 h of the reaction time over Ni/SiO2 and NC12 catalysts. The decrease in conversion might be due to the aggregation of Ni particles during the course of the reaction which is evidenced from the H2 pulse chemisorption of spent catalysts (Table 3) where we found the low dispersion and larger Ni particles. Furthermore, the drastic decrease in the yield of 2-methyltetrahyrofuran is observed over NK6 and NS16 (3D-architectures) catalysts during the time on stream after 10 h which in turn imply the confinement of Ni particles and possibly responsible for the deep hydrogenation of levulinic acid. | ||
| Fig. 9 Influence of time on stream over Ni based catalysts at 250 °C, WHSV = 1 and H2/LA = 8 (A) conversion of LA (B) yield of GVL. Reaction conditions: weight of the catalyst = 1 g, temperature = 250 °C, pressure = 1 atm, levulinic acid = 1 ml h−1, carrier gas (H2) = 1800 ml h−1, H2/LA molar ratio = 8. | ||
| Catalyst | Nm (μmol g−1) | AMSA (m2 g−1 catalyst) | AMSA (m2 g−1 Ni) | Average particle size (nm) | D (%) |
|---|---|---|---|---|---|
| NK6U | 145.76 | 11.40 | 38.01 | 17.74 | 5.70 |
| NC12U | 128.22 | 10.03 | 33.43 | 20.16 | 5.02 |
| NS16U | 185.62 | 14.52 | 48.40 | 13.93 | 7.26 |
| Ni/SiO2U | 98.06 | 7.67 | 25.57 | 26.36 | 3.84 |
It is well known that the formic acid and water both are the major unavoidable impurities in the processing of levulinic acid from the cellulosic biomass. In order to know the stability of the catalyst in the realistic condition we intentionally added the stoichiometric amount of formic acid and water to levulinic acid and carried out the reaction in optimized conditions. The reaction is carried out at optimized conditions (250 °C and 1 atm) by taking 1:1 molar ratio of levulinic acid and formic acid, in this condition formic acid, can be used as H2 source. Where we found 55, 21 and 33% of LA conversions over NK6, NC12 and NS16 respectively while the yields towards the gamma valerolactone are 28, 9 and 14. Furthermore, the reaction is carried out at optimized conditions (250 °C and 1 atm) by taking levulinic acid and water (1:1 molar ratio) along with external H2. In which we found 100, 97 and 70% of LA conversions over NK6, NC12 and NS16 respectively while the yields towards the gamma valerolactone are 71, 93 and 54%. These results suggests that the present catalytic systems are found to efficient in the realistic conditions.
| TOF of GVL = No. of moles of LA pumped per second × fractional yield of GVL/no. of surface Ni sites, |
| surface coverage (%) = AMSA (m2 g−1) × 102/SBET (m2 g−1), |
| surface density (%) = no. of surface Ni × 102/SBET (m2 g−1) |
TOF, surface coverage (%) and surface density (%) are plotted against the mesoporous silica supported Ni catalysts and displayed in Fig. 10. Since the number of surface Ni atoms are used to obtain surface density and AMSA which in turn is used to get surface coverage. Both surface density and surface coverage are proportional to each other. Even though, no. of surface Ni atoms and AMSA are proportional to Ni dispersion. Surface coverage and density need not be in proportion to Ni dispersion. This is because of variation in the BET surface area. Since catalysis is a surface phenomenon. It is more appropriate to arrive a correlation between activity and surface coverage and surface density. It can be seen from the Fig. 10 that the surface coverage (%) and surface density (%) of NC12 is greater than that of NK6 and NS16. The higher surface coverage (%) and surface density (%) of NC12 clearly indicates the presence of Ni particles located outside the pores of support (C12) while the lower surface coverage (%) and surface density (%) indicates the location of Ni particles either outside as well as inside the pore network in NK6 and NS16 catalysts. The contribution of the surface coverage and surface density (%) are higher in NC12 than those on NK6 and NS16. The drastic decrease in surface area over NK6 (Table 1) due to the presence of Ni inside the pores. As aforementioned from the TPR (Fig. 6) that the location of particles are either outside or inside the pores in case of NK6 and NS16, however in case of NC12, Ni particles are only on the outside the pores. The presence of Ni particles inside the pores has small in size than the outside the pores. The smaller particles behave differently than the bigger particles. As a result, the deep hydrogenation levulinic acid is observed in the case of NK6 and NS16.
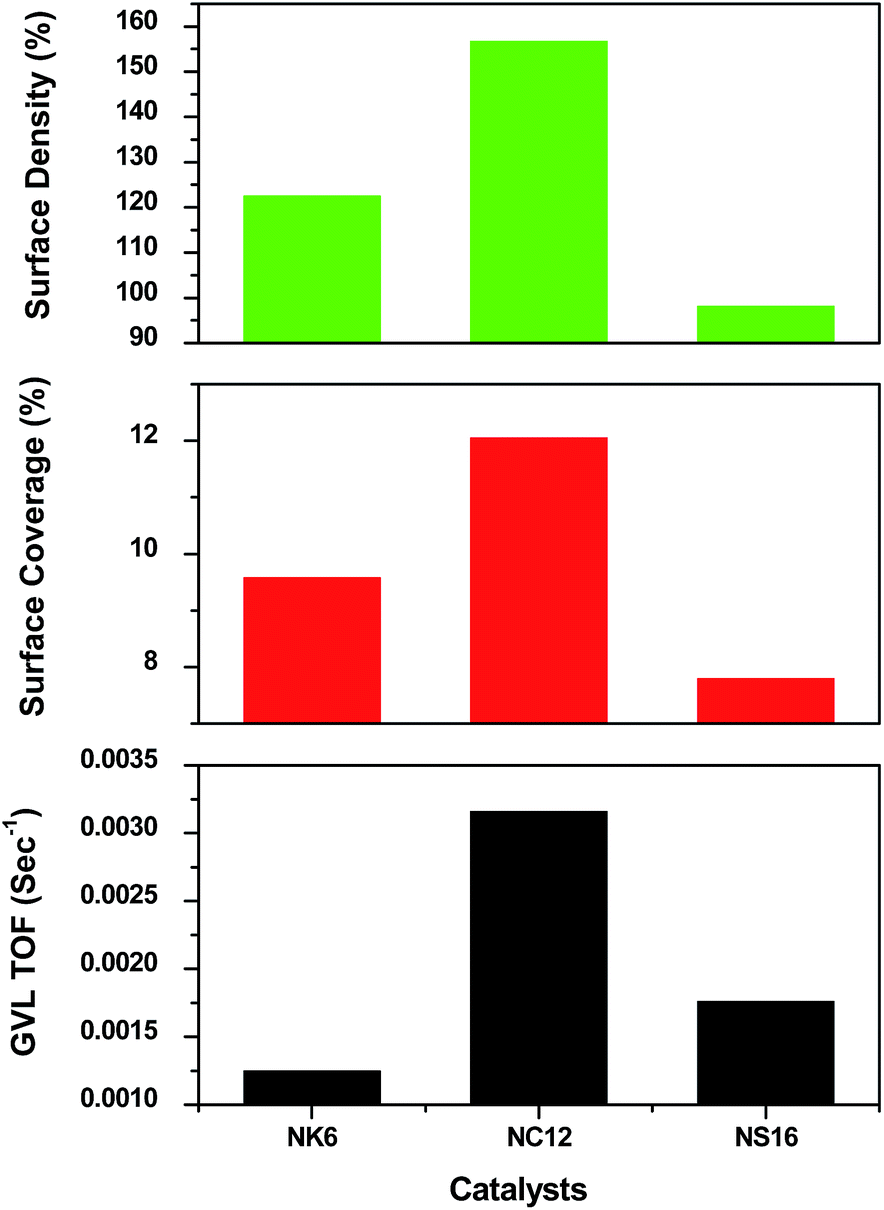 | ||
| Fig. 10 TOF, Surface coverage and surface density of mesoporous silica supported Ni catalysts. | ||
Additionally, to gain an insight into the confinement effect, the hydrogenation of γ-valerolactone has been carried out over mesoporous silicas supported Ni catalysts. The results (Fig. 11) indicate that the presence of smaller Ni particles within the pores of NK6 and NS16 yield higher conversion of γ-valerolactone (37% and 42% respectively) than that of NC12 in which Ni particles are present outside the pores (25% conversion over NC12). The formation of 2-methyl tetrahydrofuran over NK6 and NS16 is due to the confinement of Ni particles within the pores. Thus, the 3D architecture of support determines the product selectivity.
 | ||
| Fig. 11 GVL hydrogenation over mesoporous silicas supported Ni catalysts. | ||
The term, turn over frequency (TOF) is defined as the activity per site per sec. The TOF values can be obtained on the basis of LA conversion or GVL formation. Fig. 10 indicates that the TOF values towards GVL formation are in line with the surface coverage and surface density. It is more appropriate to compare the TOF values under kinetically relevant conditions. For this purpose, LA hydrogenation experiments have been carried out at higher space velocity. By using the flow rate of levulinic acid at 10 ml h−1 and the flow of H2 at 3600 ml min−1 over 1 g catalyst. The results were compiled in Table 4. From Table 4, it is noticed that the TOF of LA and GVL for NC12 catalyst is higher than that of NK6 and NS16. The higher TOF of NC12 could imply robustness of the catalytic system for the levulinic acid hydrocyclization.
| Catalysts | TOF of LA (10−4 s−1) | TOF of GVL (10−4 s−1) |
|---|---|---|
| NK6 | 12.434 | 7.22 |
| NC12 | 20.688 | 11.71 |
| NS16 | 10.64 | 5.35 |
The productivity of NC12 catalyst is calculated as described in our previous publication.19 The productivity of this catalyst for the levulinic acid hydrogenation to GVL is greater than the previously reported results and follows the trend as follows NC12 (0.9580) > Ni/HZSM-5 (0.9090)19 > Ni/SiO2 (0.8506)20 > Ni/Al2O3 (0.81055)20 > Ni/MgO (0.80079).20
Conclusions
Nickel supported on mesoporous silicas (viz, K6, C12 and S16 supports) were successfully synthesized via simple impregnation method. The conservation of a pore orderness of 2D hexagonal for C12 and the 3D cubic structures for both K6 and S16 has been observed by N2 physisorption, XRD, and TEM images. TPR and TEM results showed that relatively small size nickel nanoparticles were mainly present on the interconnected mesopores in case of K6 and S16 in addition to these particles; some of the particles were presented on the surface. Whereas in the case of C12, most of the particles were presented on the outside of the pores or on the surface. Although the conversion is same for all catalysts the product distribution is different due to the presence of small Ni nanoparticles inside the mesopores, particularly in the case of interconnected mesopore networks pertaining to enhanced Ni dispersion on NK6 and NS16. Besides, the higher activity towards GVL, NC12 catalyst is consistent during the time on stream operation up to 30 h. A slight decrease in LA conversion was observed beyond 30 h due to agglomeration of Ni particles as evidenced from the H2 pulse chemisorption of spent catalyst. A drastic decrease in LA conversion was observed over NK6 and NS16 catalysts after 10 h which might be attributed to the aggregation of Ni particles which are present in the inside the pores. These results suggested that the smaller particles agglomerate at a faster rate (NK6 and NS16) than bigger particles (NC12). Additionally, the smaller Ni particles facilitate deep hydrogenation. All these catalysts showed remarkable activity even in the presence of impurities such as formic acid and water. The present results reveal that Ni incorporated mesoporous silica catalysts are robust and are useful in the biorefinery concept. Still there is a need for the development of efficient catalysts for the direct production of 2-methyl tetrahydrofuran from levulinic acid. The further efforts are being in progress and will result in future works.Acknowledgements
The authors MV, SG, and VV gratefully acknowledge Council of Scientific and Industrial Research and University Grants Commission, New Delhi respectively for financial support.Notes and references
- BP Statistical Review of World Energy, 2011.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef
.
- P. Gullon, A. Romani, C. Vila, G. Garrote and J. C. Parajo, Biofuels, Bioprod. Biorefin., 2012, 6, 219–232 CrossRef CAS PubMed
- D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries–Industrial Processes and Products, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, Weinheim, 2006, vol. 1, p. 139 Search PubMed
- D. J. Hayes, Catal. Today, 2009, 145, 138–151 CrossRef CAS PubMed
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed. Engl., 2010, 49, 4479–4483 CrossRef CAS PubMed
- J. J. Bozell, Science, 2010, 329, 522–523 CrossRef CAS PubMed
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS PubMed
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS PubMed
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed
- K. Yamamoto, Y. Sunagawa, H. Takahashi and A. Muramatsu, Chem. Commun., 2005, 348–350 RSC
- S. B. Ren, P. Zhang, H. F. Shui, Z. P. Lei, Z. C. Wang and S. G. Kang, Catal. Commun., 2010, 12, 132–136 CrossRef CAS PubMed
- B. Xue, H. Li, J. Xu, P. Liu, Y. Zhang and Y. X. Li, Catal. Commun., 2012, 29, 153–157 CrossRef CAS PubMed
- J.-K. Lee, B.-I. An, D. Kim, S.-H. Min, J.-S. Jung and S.-H. Lee, Bull. Korean Chem. Soc., 2007, 28, 121–124 CrossRef CAS
- T. Tsoncheva, L. Ivanova, J. Rosenholm and M. Linden, Appl. Catal., B, 2009, 89, 365–374 CrossRef CAS PubMed
- K. Soni, K. C. Mouli, A. K. Dalai and J. Adjaye, Catal. Lett., 2010, 136, 116–125 CrossRef CAS
- C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636–640 CrossRef CAS
- V. Mohan, C. Raghavendra, C. V. Pramod, B. D. Raju and K. S. R. Rao, RSC Adv., 2014, 4, 9660–9668 RSC
- V. Mohan, V. Venkateshwarlu, C. V. Pramod, B. D. Raju and K. S. R. Rao, Catal.: Sci. Technol., 2014, 4, 1253–1259 CAS
- V. Mohan, B. D. Raju, K. S. R. Rao, Proceedings of International Conference on New Dimensions in Chemistry & Chemical Technologies-Applications in Pharma Industry (NDCT-2014), Spectrum Publishers, 2014, pp. 94–96, ISBN 978-93-82829-90-4 Search PubMed
- V. Mohan, B. D. Raju and K. S. R. Rao, J. Catal. Catal., 2015, 2(2), 33–38 Search PubMed
- T. W. Kim, F. Klitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601–7610 CrossRef CAS PubMed
- J. Jammaer, A. Aerts, J. D'Haen, J. W. Seo and J. A. Martens, J. Mater. Chem., 2009, 19, 8290–8293 RSC
- R. M. Grudzien, B. E. Graicka and M. Jaroniec, J. Mater. Chem., 2006, 16, 819–823 RSC
- J. R. A. Sietsma, J. D. Meeldijk, M. Versluijs-Helder, A. Broersma, A. Jos van Dillen, P. E. de Jongh and K. P. de Jong, Chem. Mater., 2008, 20, 2921–2931 CrossRef CAS
- D. Y. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS
- Z. Lihui, H. Jun, X. Songhai and L. H. Chin, Chem. Eng. J., 2007, 15, 507–511 Search PubMed
- S. Zhang, S. Muratsugu, N. Ishiguro and M. Tada, ACS Catal., 2013, 3, 1855–1864 CrossRef CAS
- H. Li, Y. Xu, H. Yang, F. Zhang and H. Li, J. Mol. Catal. A: Chem., 2009, 307, 105–114 CrossRef CAS PubMed
- K. V. R. Chary and C. S. Srikanth, Catal. Lett., 2009, 128, 164–170 CrossRef CAS PubMed
- V. Mohan, C. V. Pramod, M. Suresh, K. Hari Prasad Reddy, B. David Raju and K. S. Rama Rao, Catal. Commun., 2012, 18, 89–92 CrossRef CAS PubMed
- X. Wang, P. Wang, Z. Dong, Z. Dong, Z. Ma, J. Jiang, R. Li and J. Ma, Nanoscale Res. Lett., 2010, 5, 1468–1473 CrossRef CAS PubMed
- J. Q. Wang, L. Huang, M. Xue, Y. Wang, L. Gao, J. H. Zhu and Z. Zou, J. Phys. Chem. C, 2008, 112, 5014–5022 CAS
- F. Zhang, Y. Yan, H. Yang, Y. Meng, C. Yu, B. Tu and D. Zhao, J. Phys. Chem. B, 2005, 109, 8723–8732 CrossRef CAS PubMed
- Z. Liu, J. Zhou, K. Cao, W. Yang, H. Gao, Y. Wang and H. Li, Appl. Catal., B, 2012, 125, 324–330 CrossRef CAS PubMed
- Y. Park, T. Kang, J. Lee, P. Kim, H. Kim and J. Yi, Catal. Today, 2004, 97, 195–203 CrossRef CAS PubMed
- Q. Zhang, T. J. Wang, B. Li, T. Jiang, L. L. Ma, X. H. Zhang and Q. Y. Liu, Appl. Energy, 2012, 97, 509–513 CrossRef CAS PubMed
- D. J. Lensveld, J. G. Mesu, A. J. van Dillen and K. P. de Jong, Microporous Mesoporous Mater., 2001, 44, 401–407 CrossRef
- J. Loosdrecht, A. M. van der Kraan, A. J. van Dillen and J. W. Geus, J. Catal., 1997, 170, 217–226 CrossRef
- C. Wu, L. Wang, P. T. Williamms, J. Shi and J. Huang, Appl. Catal., B, 2011, 108–109, 6–13 CrossRef CAS PubMed
- C. Wu, L. Dong, J. Onwudili, P. T. Williams and J. Huang, ACS Sustainable Chem. Eng., 2013, 1, 1083–1091 CrossRef CAS
- R. P. Hodgkins, A. Ahniyaz, K. Parekh, L. M. Belova and L. Bergström, Langmuir, 2007, 23, 8838–8844 CrossRef CAS PubMed
- O. A. Abdelrahman, A. Heyden and J. Q. Bond, ACS Catal., 2014, 4, 1171–1181 CrossRef CAS
- R. T. Morrison and R. N. Boyd, Organic Chemistry, Allyn and Bacon, Boston, 1983, vol. 20, p. 813 Search PubMed
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10857h |
| ‡ Present address: School of Chemistry & Physics, University of KwaZulu-Natal, Westville Campus, Chiltern Hills, Durban-4000, South Africa, Tel: +27620229640; E-mail: E-mail: mohan.iict@gmail.com; , varkolum@ukzn.ac.za |
| This journal is © The Royal Society of Chemistry 2015 |