
Fast co-pyrolysis of cellulose and polypropylene using Py-GC/MS and Py-FT-IR†
Abstract
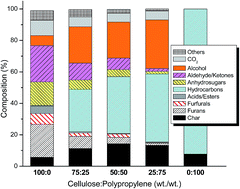
In this study, the production of high quality biofuel intermediates via fast co-pyrolysis of cellulose and polypropylene (PP) is investigated. Fast co-pyrolysis experiments were performed in a Pyroprobe® reactor and the generated vapors were analyzed using a gas chromatograph-mass spectrometer for the composition of pyrolysates, and Fourier transform infrared spectrometer for the time evolution of the key functional groups. The effects of cellulose : PP mass ratio (100 : 0, 75 : 25, 50 : 50, 25 : 75, 0 : 100) and temperature (500–800 °C) on bio-oil composition, carbon number distribution of the products, higher heating value of the products, and temporal evolution of O–H, C–O, –CH2–, CO2 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups were evaluated. Formation of long chain alcohols in the carbon number range of C8–C20 was observed as a result of the interaction of cellulose and PP. Feed composition played a decisive role in the formation of alcohols and hydrocarbons. A maximum of ca. 36% alcohols and 45% hydrocarbons were obtained from PP-rich mixture at 600 °C. The yield of char decreased and that of the aromatic hydrocarbons increased with pyrolysis temperature. Significant improvement in the heating value of the products was observed when PP was blended with cellulose. Importantly, the calculated heating values correlated well with the cumulative content of alcohols, aliphatic and aromatic hydrocarbons. The addition of PP to cellulose significantly decreased the time taken for completion of pyrolysis. Based on the product distribution, hydroxyl, hydrogen and methyl abstraction were found to be the dominant reactions involved in the transformations.
O groups were evaluated. Formation of long chain alcohols in the carbon number range of C8–C20 was observed as a result of the interaction of cellulose and PP. Feed composition played a decisive role in the formation of alcohols and hydrocarbons. A maximum of ca. 36% alcohols and 45% hydrocarbons were obtained from PP-rich mixture at 600 °C. The yield of char decreased and that of the aromatic hydrocarbons increased with pyrolysis temperature. Significant improvement in the heating value of the products was observed when PP was blended with cellulose. Importantly, the calculated heating values correlated well with the cumulative content of alcohols, aliphatic and aromatic hydrocarbons. The addition of PP to cellulose significantly decreased the time taken for completion of pyrolysis. Based on the product distribution, hydroxyl, hydrogen and methyl abstraction were found to be the dominant reactions involved in the transformations.
 Please wait while we load your content...
Please wait while we load your content...

