
Changes to amino acid composition of bloodmeal after chemical oxidation†
Abstract
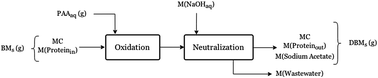
Bovine-derived bloodmeal can be decoloured using peracetic acid, extruded and injection moulded into a yellow translucent bioplastic. This plastic has different properties to extruded and injection moulded bloodmeal, in that it does not require sodium sulfite or urea to be extrudable. The effect of oxidation on the physical and chemical characteristics of the proteins during bloodmeal decolouring with PAA was studied by assessing changes in the amino acid profile. Polymer interactions, hydrophilicity and the destruction of cysteine crosslinks were measured indirectly by assessing protein solubility, molecular weight distribution and by synchrotron FT-IR analysis. Increasing peracetic acid concentration resulted in an increased loss of iron due to destruction of the porphyrin groups, increased solubility due to destruction or conversion of aromatic amino acids into hydrophilic groups, destruction of lysine, reduced protein content due to increased salt content in the final product, and a larger amount of smaller protein peptides but with a similar average molecular weight to bloodmeal. Amino acid analysis showed an increase in cysteine content in the product, FT-IR of the sulfur groups revealed that these were heavily oxidised, such that some would be unable to participate in disulfide bonds thereby increasing protein solubility.
 Please wait while we load your content...
Please wait while we load your content...

