
Design and preparation of a non-enzymatic hydrogen peroxide sensor based on a novel rigid chain liquid crystalline polymer/reduced graphene oxide composite†
Ya-Qi Yang,
He-Lou Xie*,
Jun Tang,
Shuai Tang,
Jie Yi* and
Hai-Liang Zhang*
Key Laboratory of Special Functional Polymer Materials of Hunan Province, Key Laboratory of Advanced Functional Polymer Materials of Colleges and Universities of Hunan Province and Key Lab of Environment-friendly Chemistry and Application in Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, Hunan Province, China. E-mail: xhl20040731@163.com
First published on 20th July 2015
Abstract
A novel non-enzymatic hydrogen peroxide (H2O2) sensor was developed using a rigid chain liquid crystalline (LC) polymer and reduced graphene oxide (rGO) composite. Firstly, we synthesized a novel rigid chain LC polymer with ferrocenyl as the side group. The chemical structures of the monomer and the corresponding polymer were confirmed by 1H NMR, FTIR and 1C NMR. LC behavior of the polymer was investigated by differential scanning calorimetry (DSC) and polarized light microscopy (POM). The electrochemical characterization of the PFECS/rGO films was performed by cyclic voltammetry (CV), electrochemical impedance spectroscopy (EIS), typical amperometric response (IT) measurements. The ferrocenyl in polymer presented excellent electrochemical behavior, and the modified electrode exhibited preferable electrocatalytic activity to the reduction of H2O2. The work revealed that the developed electrochemical sensor detected H2O2 with a higher sensitivity 117.142 μA mM−1 cm−2 and broader linear range between 1 × 10−5 M to 1.9 × 10−4 M.
Introduction
The fast, reliable and accurate detection of hydrogen peroxide (H2O2) has attracted considerable attention because H2O2 is a very common essential mediator in food, pharmaceutical, clinical and environmental analyses.1–4 At present, numerous methods have been developed to detect H2O2, such as fluorometry, spectrometry, chemiluminescence, chromatography and electrochemical methods.5–11 Among these methods, the electrochemical method has been extensively employed in the detection of H2O2 because of its simplicity, accurate, and fast response and economical efficiency for analysis. It is of great interest to reasonably fabricate an easy-to-make, sensitive, and cost-efficient sensor for the electrochemical method. Most of these methods are based on the immobilization of enzymes, such as horseradish peroxidase, cytochrome and myoglobin on the functionalized electrodes. Although the enzyme-based sensors have remarkable selectivity and high sensitivity for H2O2 detection, they usually suffer from complicated enzyme immobilization processes and effects of the experimental environment (pH, temperature, etc.).12 At the same time, this kind of sensor lacks enough stability originating from the intrinsic nature of the enzymes. Therefore, numerous efforts have been devoted to the development of non-enzymatic electrochemical sensors.13–15In the process of the developing the non-enzymatic electrochemical sensors, it is very important that how to increase the conductivity of the electrodes modified reasonably by the high conductive materials to improve the property of the electrochemical sensors. Graphene (Gr) is one of the most excellent candidates as the modifying materials for the electrodes because of its extra high surface area, rapid electron transport capability and thermal stability.16–18 Thus, in the past decades, numerous research has been devoted to graphene-based hybrid nanomaterials or nanocomposites for expending graphene performance.19 Furthermore, the graphene-based hybrid nanomaterial could be used to fabricate the novel mediate-free electrochemical biosensor.20 It has been proved that Gr can efficiently promote electron transfer between the redox center and the electrode surface.21
Ferrocene (Fc) and its derivatives as the most popular electrochemical active groups have been extensively used in non-enzymatic electrochemical sensors through chemically modified electrodes because of the redox reaction of Fc+/Fc is completely reversible.22 However, the adsorption of the ferrocene and its derivatives onto the electrode surface is very poor. Thus, different methods have been developed to improve the adsorption of the ferrocenes on electrode surfaces to produce chemically-modified electrodes through incorporating ferrocenes into other system.23–25 Among these methods, the typical example is that the ferrocene covalently is bound to a flexible dendritic or polymeric backbone.26 The resulting materials can be deposited onto surfaces or incorporated into carbon paste electrodes for H2O2 detection.
In this work, we designed and synthesized a rigid-chain liquid crystalline (LC) polymer (poly(2,5-bis((2-ferroceneylethyl)oxy carbonyl)styrene) (PFECS)) using Fc unit to fabricate a novel nonenzymatic H2O2 sensor combining with the reduced graphene oxide (rGO). The chemical structure is shown in Scheme 1. It is noted that in this kind of polymer the side group with two ferrocenes every unit directly links to the main chain. Generally, in this kind of polymer the significantly high steric hindrance force the flexible backbone to form rather extended and stiff, and the whole molecular chain can be considered as a cylinder because the bulky side groups located around the backbone are crowded.27,28 The typical example is mesogen-jacketed LC polymer (MJLCP).29–38 Thus, we expect the utilization efficiency of the ferrocenes would get improved because the extended and stiff main chain makes the ferrocenes more efficiently expose and contact each other. On the other hand, this kind of polymer would exhibit high electron transmission efficiency because of its every unit has two ferrocenes. At the same time, we have detailedly investigated the thermal stability, LC behavior of PFECS, and electrochemical behavior of the prepared modified electrode for detection of H2O2. The result shows that the electrode modified by PFECS/rGO film present desirable performance with good sensitivity for non-enzymatic H2O2 sensor applications.
 | ||
| Scheme 1 The chemical structure of PEECS. | ||
Experimental
Materials
Tetrahydrofuran (THF) were dried, filtered and evaporated with vacuum distillation before use. 2,2′-Azo-bis-isobutyronitrile (AIBN) was purified by recrystallization from ethanol. N,N-Dimethylformamide (DMF) (Shanghai Chemical Reagents Co., A.R. Grade), dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP) (Shanghai Chemical Reagents Co.,). Phosphate buffer solution (PBS) was prepared by mixing stock standard solution of Na2HPO4 and NaH2PO4. Stock solutions of H2O2 were freshly diluted from 30% solution (Xilong Chemical Co. Ltd). Graphite was obtained from Alfa Aesar. All other reagents were used as received from commercial sources.Instruments and measurements
1H NMR spectra were recorded on a Bruker ARX 400 MHz spectrometer with tetramethylsilane (TMS) as the internal standard and CDCl3 as the solvent. GPC measurements were carried out at 35 °C on a Waters 1515 instrument equipped with three Waters μ-Styragel columns (103, 104, and 105 Å) in series, using THF as the eluent at a flow rate of 1.0 mL min−1. The GPC data were calibrated with PS standards. Thermogravimetric analysis (TGA) was performed on a TA SDT 2960 instrument at a heating rate of 10 °C min−1 in air. Differential scanning calorimetry (DSC) experiments were carried out on a TA DSC Q100 calorimeter with a programmed heating and cooling procedure in nitrogen. The temperature and heat flow were calibrated with benzoic acid and indium. The samples were encapsulated in hermetically sealed aluminum pans with a typical sample weight of ∼5 mg. The cooling and subsequent heating DSC experiments were carried out at a rate of 10 °C min−1. LC texture of the polymers was examined under polarized optical microscope (POM, Leica DM-LM-P) coupled with a Mettler-Toledo hot stage (FP82HT). The films with a thickness of ∼10 μm were casted from THF solution on glass slides and slowly dried at room temperature.All electrochemical experiments were conducted on a CHI650D electrochemical workstation (Chenhua Instrument Company of Shanghai, China) in a conventional three-electrode system. The reference electrode was saturated calomel electrode (SCE), a platinum wire electrode was used as an auxiliary electrode and the working electrode was a modified GCE (3 mm diameter, Shanghai Chenhua, China). The pH measurements were carried out on PHS-3C exact digital pH meter (Shanghai KangYi Co. Ltd, China), which was calibrated with standard pH buffer solutions. Prior to each experiment, all solution was deoxygenated with high purity nitrogen and a nitrogen environment was then kept over the solution in the cell. All experiments were carried out at room temperature.
Experimental section
Synthesis
The synthetic route of PEECS and the corresponding monomer are shown in Scheme 2. And the detail information about the synthesis and character of the intermediate and monomers are presented as following. | ||
| Scheme 2 Synthetic route of the monomer and the corresponding polymer. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 5.75 (d, 1H, CH2), 5.42 (d, 1H, CH2).CH–), 5.44 (d, 1H, CH2), 5.41 (d, 1H, CH2), 4.48 (m, 2H, OCH2), 4.15 (br s, 9H, Cp), 2.81 (t, J = 6.5 Hz, 2H, OCH2CH2). IR (KBr, cm−1): ν 3090 (w), 2960 (w), 2845 (w), 1561 (w), 1482 (w), 1452 (w), 1295 (m), 1239 (s), 1186 (m), 1107 (s), 1057 (m), 994 (m), 921 (m), 821 (m), 797 (s), 759 (s), 718 (m). 13C NMR (CDCl3) δ (ppm): 29.12 (–OCH2
CH–), 5.75 (d, 1H, CH2), 5.42 (d, 1H, CH2).CH–), 5.44 (d, 1H, CH2), 5.41 (d, 1H, CH2), 4.48 (m, 2H, OCH2), 4.15 (br s, 9H, Cp), 2.81 (t, J = 6.5 Hz, 2H, OCH2CH2). IR (KBr, cm−1): ν 3090 (w), 2960 (w), 2845 (w), 1561 (w), 1482 (w), 1452 (w), 1295 (m), 1239 (s), 1186 (m), 1107 (s), 1057 (m), 994 (m), 921 (m), 821 (m), 797 (s), 759 (s), 718 (m). 13C NMR (CDCl3) δ (ppm): 29.12 (–OCH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2), 66.05–75.4 (Cp–), 68.7 (–OH2CH2), 117.8 (H2), 128.15 (aromatic ortho to C–CHCH2), 128.36 (––CHCH2), 130.45 (aromatic ortho to C–CO), 132.51 (aromatic ortho to C–CO), 133.33 (–CO), 134.95 (–HCH2), 139.56 (–CO), 165.54 (O), 166.74 (O).
H2), 66.05–75.4 (Cp–), 68.7 (–OH2CH2), 117.8 (H2), 128.15 (aromatic ortho to C–CHCH2), 128.36 (––CHCH2), 130.45 (aromatic ortho to C–CO), 132.51 (aromatic ortho to C–CO), 133.33 (–CO), 134.95 (–HCH2), 139.56 (–CO), 165.54 (O), 166.74 (O).Preparation of the PFECS/rGO modified electrode
The graphite oxide (GO) powder was prepared from graphite powder via a modified Hummers' method. The surface of electrode was polished successively with 0.3, 0.1 and 0.05 μm alumina slurry and then cleaned under ultra sonication in ethanol and water, respectively. Then, 5 μL the GO solution was casted on the surface of glassy carbon electrode (GCE) and allowed to dry at ambient temperature. Cyclic voltammograms (CV) was used to reduce GO at a potential range from 0 V to −1.4 V in 0.02 M pH = 5.6 phosphate buffer solution with N2-saturated. In this work, 40 cycles were adopted for the reductive adsorption of GO on the electrode. While the tiled graphene modified electrode was obtained. The graphene modified electrodes were then dried under ambient temperature. 5.0 mg PFECS were dispersed in 5.0 mL DCM. Then 5 μL of 5.0 mg mL−1 PFECS solution and equivalent volume of the resulting mixture were dropped onto the rGO modified electrode surface successively and dried in room temperature. The modified electrodes were stored at 4 °C in a refrigerator.Results and discussion
Synthesis and characterization of PFECS
The objective polymer was readily obtained by radical polymerization method because the monomer has vinyl substituent. Fig. 1 depicts the 1H NMR spectra of the monomer and the polymer, wherein the assignments of resonance peaks are included. The characteristic resonance peaks of the vinyl substituent of corresponding monomer FECS at chemical shifts of 5.78, 5.74, 5.44, and 5.41 ppm (denoted as e and f, respectively) disappeared completely after polymerization. Moreover, the chemical shifts of PFECS became quite broad compared to the monomer FECS, which was consistent with the polymer structure expected. Generally, the formation of film and was strongly depended on the molecular weight (MW), and the kind of LC polymer presented LC behavior only its MW exceeded a critical value. Thus, we furthermore used GPC to characterize the MW and polymer polydispersity index (PDI). The result showed its MW was 6.0 × 104 and the PDI was 1.83. According to the previous results, the present MW was big enough to prepare film and present LC behavior. | ||
| Fig. 1 1H NMR spectra of FECS and the PFECS. | ||
Thermal and LC properties of the PFECS
In present system, the annealing process was beneficial to the development of LC phase and film formation, which was important for the chemical or electrochemical property. Thus, we firstly investigated the thermal stability of PFECS. TGA experimental results revealed that the thermal decomposition temperature of the PFECS was above 340 °C, indicating that this kind of polymer had good thermal stabilities.To investigate the liquid-crystalline behavior of PFECS, the combined techniques of DSC and POM were employed to use. The sample was completely dried under vacuum before performing the DSC analysis. To eliminate the effect of thermal history, the sample was heated from room temperature to 150 °C with a rate of 10 °C min−1, kept at 150 °C for 5 min and then cooled to 0 °C at a rate of 10 °C min−1. Fig. 2(a) describes of DSC heating traces of the PFECS recorded at a rate of 10 °C min−1 after erasing the sample's thermal history. The DSC measurements showed only a glass transition step at about 93 °C but no endothermic peak correlative to any melting of crystals. This result was the same to the traditional MJLCPs.41
 | ||
| Fig. 2 (a) DSC curves of the polymer PFECS, (b–d) POM micrographs of PFECS (magnification 200×). | ||
Before the POM experiment, the sample was casted from THF solution and slowly dried at room temperature. The POM micrographs of the polymer were recorded during the first cooling process. The experimental phenomenon showed the samples presented birefringence under different temperature. Even cooling to room temperature, the birefringence still remained. All results revealed the sample formed stable LC phase under different temperature.42 Obviously, the kind of polymer didn't include LC mesogens. Thus, the LC phenomenon could be explained by the chain conformation, that was, the bulky side group constituted of two ferrocenes and one benzene located around the backbone were crowded, and the significantly high steric hindrance forced the flexible polyolefin backbone to become rather extended and stiff, and the whole molecular chain would be considered as a molecule, furthermore, the LC molecule self assembly into LC phase, which was similar to the previous results reported in the literature.43
Direct electrochemical of PFECS/rGO modified electrode
Fig. 3 shows the CV of different electrodes modified in deaerated 0.2 M PBS (pH 7.0). For PFECS/GCE, a pair of small redox peaks consisted of an anodic peak at 0.526 V and a cathodic peak at 0.402 V was observed. Compared with electrode modified by PFECS, the peak currents of electrode modified by PFECS/rGO increased obviously, and a pair of well-defined redox peaks was observed. The anodic peak potential (Epa) and cathodic peak potential (Epc) were located at 0.535 V and 0.462 V, respectively. The formal potential (defined as the midpoint of reduction and oxidation potentials), E0′ was 0.486 V for PFECS. The peak potential separation (Ep, defined as Ep = Epa − Epc) was calculated as 73 mV. All the above results revealed that the direct electron transfer between PFECS and electrode was greatly enhanced. | ||
| Fig. 3 The CV of different materials modified electrodes in 0.2 M PBS at a scan rate of 100 mV s−1. | ||
The film conductivity in the presence of rGO was studied by electrochemical impedance spectroscopy (EIS). Fig. 4 describes the electrochemical impedance spectra of different modified electrode. The electron transfer resistance (Ret) at the electrode surface could be used to describe the interface properties of the electrode, which was equal to the semicircle diameter of electrochemical impedance spectra. For electrochemical impedance spectrum of GCE, the semicircle domain was very small (Ret = 1338 Ω), which indicated very low electron transfer resistance to the redox-probe dissolved in the electrolyte solution. However, the Ret (2681 Ω) of the PFECS modified electrode was much larger than that of the GCE. It implied that PFECS film on the electrode surface could inhibit the electron transfer of the redox-probe of [Fe(CN)6]P3−/4− to the electrode surface to a certain degree. After added rGO, the Ret of PFECS/rGO modified electrode decreased to about 302 Ω, indicating that rGO had good conductivity and the PFECS/rGO film could make the electron transfer more easily.
 | ||
| Fig. 4 Electrochemical impedance spectra of different modified electrode at the open circuit potential in 0.2 M PBS, pH = 7.0, containing 5 mM K3Fe(CN)6 and 5 mM K4Fe(CN)6. | ||
The CVs of PFECS/rGO modified electrode with different scan rates were measured as shown in Fig. 5. A series of well-defined redox peaks shape was observed with different scan rates in the range of 0.05–0.15 V s−1, which showed almost equal heights of reduction and oxidation peaks at the same scan rate. As seen in Fig. 5(b), the anodic and cathodic peak current for PFECS/rGO modified electrode exhibits a linear relationship with the scan rate, indicating a typical surface-controlled electrode process.
 | ||
| Fig. 5 (a) CVs of PFECS/rGO/GCE in PBS (pH 7.0) at different scan rates. (b) Plot of the peak current i against the scan rate v. | ||
Electrochemical response to H2O2 at the PFECS/rGO modified electrode
Electroanalytical method of the PFECS/rGO film electrode could be applied in the analysis of H2O2. Fig. 6 shows the cyclic voltammograms of the PFECS/rGO modified electrode in 0.2 M pH 7.0 PBS in the absence and presence of different amount of H2O2 at 0.1 V s−1. When H2O2 was added into the blank buffer, a dramatic change in the cyclic voltammogram occurred with an increase in cathodic current. The catalytic reduction occurred at potential of 0.4 V at the modified electrode. A significant improvement of the cathodic current reflected the high electrocatalytic activity of the PFECS/rGO modified electrode. The mechanism of electrocatalytic reduction of H2O2 at PFECS/rGO film electrode is given in Scheme 3. The ferrocenyl in PFECS which was in the oxidised form at the electrode surface gets reduced at the applied potential. This reduced form of the mediator in turn reduces H2O2 in solution and gets oxidised. The regenerated oxidised form of PFECS at the surface of the electrode again got reduced producing the reduction current at the same time. The process of analyte reduction and catalyst regeneration proceeds in a cyclic manner enhancing the reduction current of the mediator.44 | ||
| Fig. 6 CVs of PFECS/rGO in 0.2 M PBS (pH 7.0) in the presence and absence of H2O2. | ||
 | ||
| Scheme 3 Mechanism of H2O2 reduction with PFECS/rGO. | ||
The PFECS/rGO modified electrode could also be used as an amperometric sensor for the detection of H2O2. Typical current–time (i–t) curves for the modified electrodes (PFECS/rGO) with successive addition of H2O2 at a working potential of 0.44 V (V vs. Ag/AgCl) are presented in Fig. 7. When H2O2 solution was continuously injected into the PBS solution, the current was increased and then reached to steady with a response time lower than 3 s, indicating a fast response. This phenomenon was mainly due to the existence of Fc and the well-conductive properties of the PFECS. At the same time, a shift of peak potential could also be observed in Fig. 6, where the explanation was expecting for further research. Fig. 7 shows the change in the response current with the concentration of H2O2. The reduction currents were proportional to the concentration of H2O2 ranging from 1.0 × 10−5 to 1.9 × 10−4 M, with a regression equation of Ip (μA) = −0.0082C − 0.06824 (μM) (R2 = 0.999). The sensitivity of the PFECS/rGO sensor was calculated as 117.142 μA mM−1 cm−2 by dividing the slope of the linear regression equation by the electrode surface area.
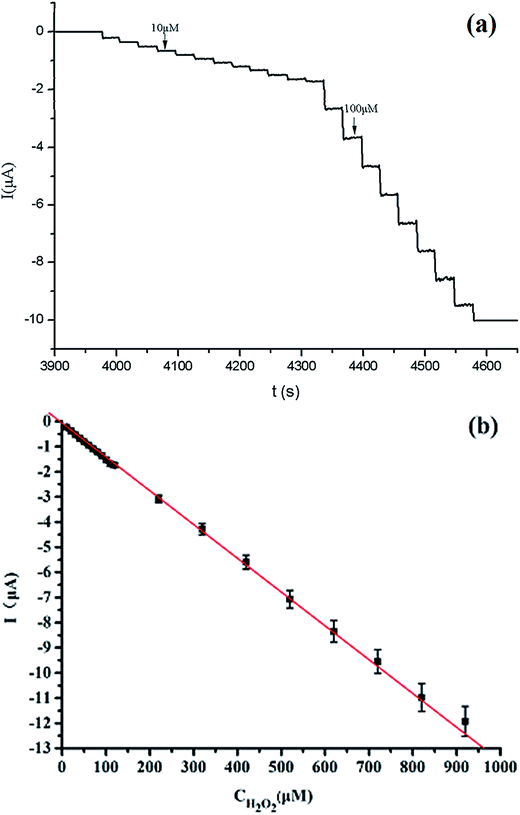 | ||
| Fig. 7 (a) Amperometric response of the H2O2 sensor to successive addition of different concentration of H2O2 in PBS solution (pH = 7.0) at the working potential of 0.44 V. (b) Plot of electrocatalytic peak current vs. concentration of H2O2. | ||
A comparison of this PFECS/rGO/GCE with other H2O2 sensors reported in the literature are shown in Table 1 in terms of linear detection range (LDR), limit of detection (LOD) and sensitivity. The analytical performances for PFECS/rGO/GCE are comparable and better than those obtained on other electrodes reported recently. Therefore, the PFECS/rGO/GCE can be used as a non-enzyme platform for the effective detection of H2O2 with low LOD and high sensitivity. It indicates in Table 1 that the limit of detection of the sensor is relative lower than previous reports. Meanwhile, in order to test the repeatability, reproducibility and re-usability of the sensor, the relative standard deviation (RSD) and the response current depended on the time are shown in Fig. 1 and 2 in the ESI,† indicating the PFECS/rGO/GCE possessed acceptable reproducibility and stability. Moreover, the possible interferences from some co-existing electroactive compounds were assessed in order to investigate the selectivity of the developed non-enzymatic sensor as show in Fig. 3 of the ESI.† It reveals that the proposed non-enzymatic electrode can be employed in the selective detection of H2O2 in the presence of these common physiological materials.
Obviously, the result indicated that this novel composites PFECS/rGO presented excellent electrochemical properties, which could be used for the detection of H2O2. Compared to the other polymer, the whole molecular chain of the polymer PFECS would be considered as a cylinder because the significantly high steric hindrance force the flexible backbone to become rather extended and stiff. At the same time, the side group with two ferrocenes every unit which was helpful to high electron transmission efficiency was located around the backbone. We speculated the stiff main chain and high density electronic sites may make the ferrocenes more efficiently expose and contact each other, which may be an new strategy for molecular design using sensor's material. According to this idea, the relative optimization results will report in the following work.
Conclusions
The monomers 2,5-bis[(2-ferroceneylethyl)oxy carbonyl]styrene were successfully synthesized and the corresponding polymer was easily obtained via radical polymerization. The chemical structure of the polymer was confirmed by various characterization techniques. The polymer showed good thermostability and birefringent melts. Furthermore, based on a new PFECS/rGO composite film, a novel amperometric H2O2 sensor was designed. The rGO have improved the conductivity and mechanical properties of the PFECS. Combining the advantages of PFECS and rGO the modified electrode showed good analytical performance, such as high sensitivity and storage stability. Hence, it provides a promising application for developing nonenzymatic sensor with transition metal and rigid chain LC polymers materials.Acknowledgements
This research is financially supported by the National Nature Science Foundation of China (21374092 and 21104062), the project of Hunan Provincial Education Department (YB2013B032), Innovation Platform Open Foundation of University of Hunan Province (14K093) and Beijing National Laboratory for Molecular Sciences (BNLMS).Notes and references
- N. Akhtar, A. M. Khairy and A. El-Saida, Sens. Actuators, B, 2015, 207, 158 CrossRef CAS PubMed.
- C. Y. An, K. Zhou, W. J. Kim and C. H. Chung, Sens. Actuators, B, 2015, 213, 329 CrossRef CAS PubMed.
- Y. C. Li, Y. Y. Zhang, Y. M. Zhong and S. X. Li, Appl. Surf. Sci., 2015, 347, 428 CrossRef CAS PubMed.
- Q. W. Liu, T. Zhang, L. L. Yu, N. Q. Jia and D. P. Yang, Analyst, 2013, 138, 5559 RSC.
- G. B. Xu and S. J. Dong, Electroanalysis, 1999, 11, 1180 CrossRef CAS.
- N. Masuoka, M. Wakimoto, T. Ubuka and T. Nakano, Clin. Chim. Acta, 1996, 254, 101 CrossRef CAS.
- F. J. Pérez and S. Rubio, Plant Growth Regul., 2006, 48, 89 CrossRef.
- Y. Z. Li and A. Townshend, Anal. Chim. Acta, 1998, 359, 149 CrossRef CAS.
- R. Schulte-Ladbeck, P. Kolla and U. Karst, Anal. Chem., 2003, 75, 731 CrossRef CAS.
- F. Li, W. Chen, C. F. Tang and S. S. Zhang, Talanta, 2009, 77, 1304 CrossRef CAS PubMed.
- X. P. Lu, X. P. Xiao, Z. Li, F. G. Xu, H. L. Tan, L. L. Sun and L. Wang, Anal. Methods, 2014, 6, 235 RSC.
- X. Y. Li, Y. X. Liu, L. C. Zheng, M. J. Dong, Z. H. Xue, X. Q. Lu and X. H. Liu, Electrochim. Acta, 2013, 113, 170 CrossRef CAS PubMed.
- S. J. He, Z. G. Chen, Y. Y. Yu and L. J. Shi, RSC Adv., 2014, 4, 45185 RSC.
- D. G. James, A. S. Karakoti, T. Inerbaev, S. Sanghavi, P. Nachimuthu, V. Shutthanandan, S. Seal and S. Thevuthasan, J. Mater. Chem. B, 2013, 1, 3443 RSC.
- H. J. Liu, D. W. Yang and H. H. Liu, Anal. Methods, 2012, 4, 1421 RSC.
- S. L. Yang, G. Li, G. F. Wang, J. H. Zhao, M. F. Zhao and L. B. Qu, Sens. Actuators, B, 2015, 208, 593 CrossRef CAS PubMed.
- F. Lorestani, Z. Shanhnavaz, P. Mn, Y. Alias and N. S. A. Manan, Sens. Actuators, B, 2015, 208, 389 CrossRef CAS PubMed.
- P. P. Liu, J. W. Li, X. H. Liu, M. Li and X. Q. Lu, J. Electroanal. Chem., 2015, 751, 1 CrossRef CAS PubMed.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- P. F. Pang, Z. M. Yang, S. X. Xiao, J. L. Xie, Y. L. Zhang and Y. T. Gao, J. Electroanal. Chem., 2014, 727, 27 CrossRef CAS PubMed.
- Y. P. Ye, T. Kong, X. F. Yu, Y. K. Wu, K. Zhang and X. P. Wang, Talanta, 2012, 89, 417 CrossRef CAS PubMed.
- G. C. Zhao, M. Q. Xu and Q. Zhang, Electrochem. Commun., 2008, 10, 1924 CrossRef CAS PubMed.
- T. Ghosh, P. Sarkar and A. P. F. Turner, Bioelectrochemistry, 2015, 102, 1 CrossRef CAS PubMed.
- T. Li and M. H. Yang, Sens. Actuators, B, 2011, 158, 361 CrossRef CAS PubMed.
- Y. Z. Lu, Y. Y. Jiang, H. B. Wu and W. Chen, Electrochim. Acta, 2015, 156, 267 CrossRef CAS PubMed.
- R. K. Nagarale, J. M. Lee and W. Shin, Electrochim. Acta, 2009, 54, 6508 CrossRef CAS PubMed.
- J. J. Watanabe, N. Sekine, T. Nematsu and M. Sone, Macromolecules, 1996, 29, 4816 CrossRef CAS.
- B. M. Rosen, C. J. Wilson, D. A. Wilson, M. Peterca, R. Imam and V. Percec, Chem. Rev., 2009, 109, 6275 CrossRef CAS PubMed.
- Q. F. Zhou, A. M. Li and X. D. Feng, Macromolecules, 1987, 20, 233 CrossRef CAS.
- L. Weng, J. J. Yan, H. L. Xie, G. Q. Zhong, S. Q. Zhu, H. L. Zhang and E. Q. Chen, J. Polym. Sci., Part A-1: Polym. Chem., 2013, 51, 1912 CrossRef CAS PubMed.
- G. H. Wen, B. Zhang, H. L. Xie, X. Liu, G. Q. Zhong, H. L. Zhang and E. Q. Chen, Macromolecules, 2013, 46, 5249 CrossRef CAS.
- H. L. Xie, S. J. Wang, G. Q. Zhong, Y. X. Liu, H. L. Zhang and E. Q. Chen, Macromolecules, 2011, 44, 7600 CrossRef CAS.
- H. L. Xie, C. K. Jie, Z. Q. Yu, X. B. Liu, H. L. Zhang, Z. H. Shen, E. Q. Chen and Q. F. Zhou, J. Am. Chem. Soc., 2010, 132, 8071 CrossRef CAS PubMed.
- H. L. Xie, Y. X. Liu, G. Q. Zhong, H. L. Zhang, E. Q. Chen and Q. F. Zhou, Macromolecules, 2009, 42, 8774 CrossRef CAS.
- D. Wei, A. Brûlet, P. A. Albouy, P. Keller, X. G. Wang and M. H. Li, Chin. J. Polym. Sci., 2012, 30, 258 CrossRef.
- B. W. Qiu, F. Chen, Y. G. Shangguan, Y. Lin and Q. Zheng, Chin. J. Polym. Sci., 2015, 33, 95 CrossRef CAS.
- X. B. Liu, Y. F. Zhu, H. Fan and E. Q. Chen, Chin. J. Polym. Sci., 2013, 31, 946 CrossRef CAS.
- Z. Q. Yu, J. W. Y. Lam, K. Q. Zhao, C. Z. Zhu, S. Yang, J. S. Lin, B. S. Li, J. H. Liu, E. Q. Chen and B. Z. Tang, Polym. Chem., 2013, 4, 996 RSC.
- Y. L. Yang, Z. W. Xie and C. Wu, Macromolecules, 2002, 35, 3426 CrossRef CAS.
- D. Zhang, Y. X. Liu, X. H. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 4494 CrossRef CAS.
- D. Zhang, Y. X. Liu, X. H. Wan and Q. F. Zhou, Macromolecules, 1999, 32, 5183 CrossRef CAS.
- S. P. Hendry, M. F. Cardosi, A. P. F. Turner and E. W. Neuse, Anal. Chim. Acta, 1993, 281, 453 CrossRef CAS.
- W. A. Amer, L. Wang, A. M. Amin, H. J. Yu, L. Zhang, C. Li and Y. Wang, Polym. Adv. Technol., 2013, 24, 181 CrossRef CAS PubMed.
- M. P. G. Armada, J. Losada, I. Cuadrado, B. Alonso, B. González, C. M. Casado and J. B. Zhang, Sens. Actuators, B, 2004, 101, 143 CrossRef PubMed.
- X. J. Bian, X. F. Lu, E. Jin, L. R. Kong, W. J. Zhang and C. Wang, Talanta, 2010, 81, 813 CrossRef CAS PubMed.
- J. G. Hu, F. H. Li, K. K. Wang, D. X. Han, Q. X. Zhang, J. H. Yuan and L. Niu, Talanta, 2012, 93, 345 CrossRef CAS PubMed.
- M. R. Mahmoudian, Y. Alias, W. J. Basirun, A. M. Golsheikh and F. Jamali-Sheini, Mater. Chem. Phys., 2013, 141, 298 CrossRef CAS PubMed.
- E. Jin, X. F. Lu, L. L. Cui, D. M. Chao and C. Wang, Electrochim. Acta, 2010, 55, 7230 CrossRef CAS PubMed.
- P. Gao, Y. X. Gong, N. P. Mellott and D. W. Liu, Electrochim. Acta, 2015, 173, 31 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10540d |
| This journal is © The Royal Society of Chemistry 2015 |