
Deoxygenation of graphene oxide using household baking soda as a reducing agent: a green approach
Abstract
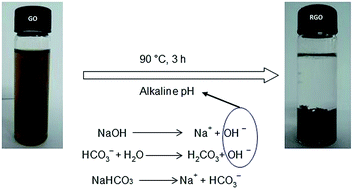
A one-step, novel, easy, fast, facile, economic, and environmental friendly route to reduce graphene oxide (GO) is studied and explained in this study. The household baking soda (sodium bicarbonate/NaHCO3) was applied here as a reducing agent. In aqueous solution, NaHCO3 hydrolyses into hydroxide ion and leads to deoxygenate GO sheets. The confirmation of oxygen reduction was checked with UV-visible spectroscopy, X-ray diffraction, Fourier transform infrared spectrometry and Raman spectroscopy. Thermal behavior of graphene was analyzed by thermogravimetry analysis and differential scanning calorimetry. Again, atomic force microscopy, scanning electron microscopy, field emission scanning electron microscopy and transmission electron microscopy were used for morphological study. Energy dispersive X-ray spectrometer was applied to analyze the elemental composition. The results of the above experiments demonstrate that the house-hold baking soda is able to produce functional graphene, which can be a suitable replacement of hydrazine, sodium borohydrate or other toxic reducing agents. This method is high yielding and safe to use in biomaterial applications.
 Please wait while we load your content...
Please wait while we load your content...

