DOI:
10.1039/C5RA10428A
(Paper)
RSC Adv., 2015,
5, 57472-57481
Metal-free regioselective C-3 acetoxylation of N-substituted indoles: crucial impact of nitrogen-substituent†
Received
2nd June 2015
, Accepted 24th June 2015
First published on 24th June 2015
Abstract
A metal-free method for the regioselective C-3 acetoxylation of the N-substituted indoles with PhI(OAc)2 is described under mild reaction conditions. This method tolerates a broad range of functional groups with moderate to good yields. The π-electron-deficient aryl-substituents on the N-atom of indoles and the acidic reaction medium remarkably favor C-3 acetoxylation.
Introduction
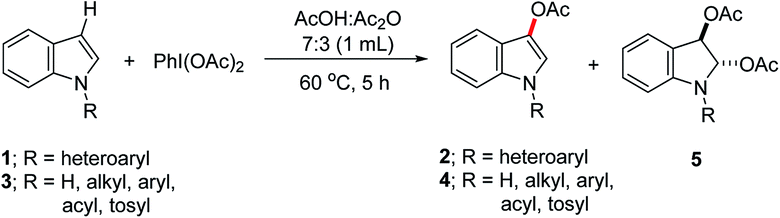
Direct functionalization of a C–H bond to form a C–O bond is amongst the most demanding chemical reactions in synthetic organic chemistry.1 The C–O bond formation in arenes to obtain hydroxylated-,2 alkoxylated-3 or acetoxylated-arenes4 have been extensively explored, and regioselectivity has been successfully achieved by the installation of a Lewis-base directing group. In contrast to many reports on the acetoxylation of arenes, the synthesis of the regioselectively acetoxylated heteroarenes is relatively scarce.5 Among the heteroarenes, the indole derivatives are very important building blocks, found in many pharmaceuticals as well as in the biologically active compounds. In particular, 3-acetoxyindoles are potential materials, applied for the detection of acetylcholinesterase in tissue slices6 and they could also be used as starting precursors for the synthesis of potential 5-hydroxytryptamine6 (5-HT6) receptor ligand scaffolds.7 Hence, the efficient synthesis of 3-acetoxyindoles through the regioselective C-3 acetoxylation of indole derivatives is highly desirable. The groups of Lei,8 Kwong7 and Suna9 have independently reported the C-3 acetoxylation of N-substituted indoles using Pd(OAc)2 catalyst, and PhI(OAc)2 as external oxidant (Scheme 1a). This palladium-catalyzed regioselective acetoxylation process is applicable to the various indole substrates containing methyl, benzyl, aryl or arenesulfonyl groups as the nitrogen-substituents. However, the employment of the precious metal catalysts for the functionalization of probable pharmacological indole derivatives, limit the advancement of these metal-catalyzed acetoxylation processes.
 |
| | Scheme 1 Regioselective C-3 acetoxylation of substituted-indoles: (a) palladium-catalyzed acetoxylation of N-methyl, N-benzyl- or N-aryl indoles, (b) this work demonstrates: metal-free acetoxylation of N-(hetero)aryl indoles. | |
In recent years, numerous reports have been described for the C–H bond functionalization under the transition-metal-free conditions.10 A transition-metal-free C-3 acetoxylation of the free NH-indole has been described under the strong basic condition by Huang and co-workers.11 However, this method is not applicable to the N-substituted indole substrates and has limited scope. The C-3 acetoxylation of N-Boc-protected indole has been observed by Lei et al. during their studies directed toward the synthesis of diacetoxylated indolines.12 To our knowledge, a comprehensive approach for the metal-free C-3 acetoxylation of the N-substituted indoles with high regioselectivity is not known. Herein, we present the regioselective C-3 acetoxylation of the N-substituted indoles with PhI(OAc)2 as an oxidant under the metal-free and mild reaction conditions. Moreover, we have demonstrated the effect of the N-substituents on reactivity and selectivity of the indole acetoxylations.
Results and discussion
During the investigation of a less expensive first-row transition metal catalyst for the acetoxylation of the N-substituted indoles employing PhI(OAc)2 in acetic acid solvent, we observed that the C-3 acetoxylation of indole occurred in the absence of metal catalysts, when a suitable substituent on the nitrogen atom of indole was installed. For example, the reaction of 1-pyrimidinyl indole (1a) with PhI(OAc)2 in AcOH/Ac2O solvent at 60 °C, exclusively produced 1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2a) in 93% isolated yield (Table 1, entry 1). The 1-arylindoles, like 1-phenylindole (3a) reacted with PhI(OAc)2 under the metal-free condition to afford the corresponding C-3 acetoxylated indole 4a in 50% yield; whereas the reaction of 1-(4-methoxyphenyl)indole (3b) with PhI(OAc)2 under the same condition produced only 37% of the acetoxylated compound 4b. Surprisingly, the free NH-indole or indoles having electron-rich substituents at the nitrogen-center, such as 1-methylindole, were decomposed to the unknown compounds at room temperature. However, the indoles bearing strong electron-withdrawing substituents at the nitrogen-atom, such as 1-(acetyl)indole and 1-(tosyl)indole produced the diacetoxylated indolines in 91% and 28% yields, respectively (Table 1, entries 6 and 7).12 These studies on the effect of N-substituents towards the acetoxylation of indoles suggested that a π-electron-deficient (hetero)arene-substituent on the nitrogen atom of indole greatly enhances the selective C-3 acetoxylation reaction.
Table 1 Effect of nitrogen-substituents on C-3 acetoxylation of indolesa
|
| Entry |
R |
% Yield (2 or 4) |
5 |
Recovered (1 or 3) |
| Reaction conditions: compound 1 or 3 (0.30 mmol), PhI(OAc)2 (0.106 g, 0.33 mmol), AcOH/Ac2O (1.0 mL), yields of isolated compounds. Starting compound was not recovered. Decomposed into intractable products at room temperature. |
| 1 |
2-Pym |
93 (2a) |
— |
— |
| 2 |
Ph |
50 (4a) |
— |
35 (3a) |
| 3 |
4-MeO-C6H4 |
37 (4b) |
— |
—b |
| 4 |
H |
— |
— |
—c |
| 5 |
CH3 |
— |
— |
—c |
| 6 |
C(O)CH3 |
— |
91 |
5 |
| 7 |
Tosyl |
— |
28 |
70 |
After optimizing the effect of N-substituent's on the metal-free acetoxylation of indoles, we have tested the impact of various solvents and oxidants on the acetoxylation reaction (Table 2). The use of glacial acetic acid as solvent instead of AcOH/Ac2O, produced the desired acetoxylated product 2a in 76% isolated yield (entry 2). However, the complete decomposition of 1a was observed in TFA/TFAA under the reaction conditions (entry 3). The employment of non-acid solvent 2,2,2-trifluoroethanol (TFE) gave only 20% of product 2a, whereas the reactions in CH3CN or 1,2-dichloroethane (DCE) did not produce any acetoxylated products (entry 4 and 5). The presence of strong oxidant PhI(TFA)2 resulted in the decomposition of 1a; however, other organic oxidants, such as m-CPBA, tert-BuOOH or inorganic oxidants like K2S2O8 and oxone were ineffective, and most of the starting material was recovered (entry 6–8).
Table 2 Optimization of reaction parameters for C-3 acetoxylation of 1aa

|
| Entry |
Variation from “standard condition” |
Yieldb (%) |
| Reaction conditions: 1a (0.059 g, 0.30 mmol), PhI(OAc)2 (0.106 g, 0.33 mmol), AcOH/Ac2O (1.0 mL). TFE (2,2,2-trifluoroethanol), TFA (trifluoroacetic acid), TFAA (trifluoroacetic anhydride), NR = no reaction. Yields of isolated compounds. Decomposed into intractable compounds. |
| 1 |
— |
93 |
| 2 |
AcOH instead of AcOH/Ac2O |
76 |
| 3 |
TFA/TFAA instead of AcOH/Ac2O |
—c |
| 4 |
TFE instead of AcOH/Ac2O |
20 |
| 5 |
CH3CN, DCE instead of AcOH/Ac2O |
NR |
| 6 |
PhI(TFA)2 instead of PhI(OAc)2 |
—c |
| 7 |
m-CPBA instead of PhI(OAc)2 |
Trace |
| 8 |
K2S2O8, oxone or t-BuOOH instead of PhI(OAc)2 |
NR |
| 9 |
25 °C instead of 60 °C |
15 |
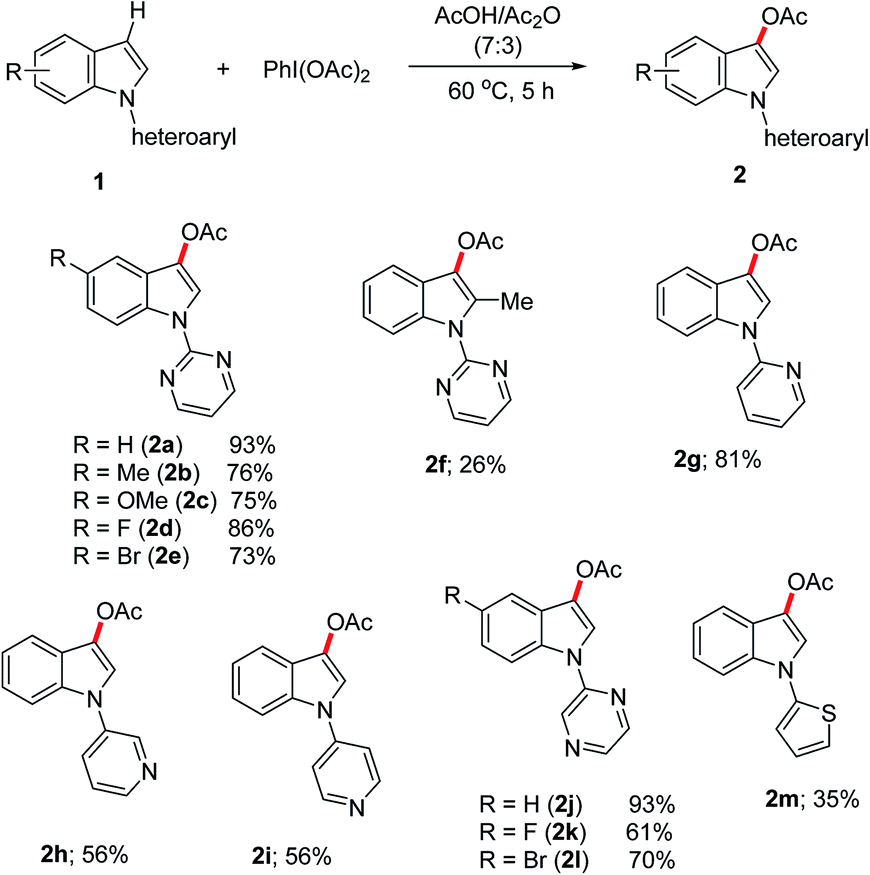
Having the optimized reaction conditions in hand, we probed the scope of the metal-free C-3 acetoxylation of the N-heteroaryl-substituted indoles 1 employing PhI(OAc)2 as the oxidant in AcOH/Ac2O solvent (Scheme 2). Notably, the N-pyrimidinyl indoles containing electronically different functional groups reacted smoothly to produce the desired acetoxylated products (2a–2e) in good yields. The tolerability of the functional groups such as –F, –Br is significant, as they can be further functionalized into important compounds. It is noteworthy that the direct acetoxylation of indoles 1 occurred with excellent regioselectivity to predominantly produce C-3 acetoxylated indoles. Particularly, by employing 2-pyrimidinyl as the N-substituent on indoles, neither the starting compounds nor the acetoxylated products were decomposed. More hindered 2-substituted indole 1f reacted with low efficacy to produced 2f in 26% yield. In addition to the N-pyrimidinyl indoles, the indoles containing 2-pyridinyl and 2-pyrazinyl as nitrogen-substituents reacted efficiently to give the desired acetoxylated products in good yields. The pyridinyl-substituted indoles, 1h and 1i reacted moderately to yield the corresponding acetoxylated compounds 2h and 2i, respectively. Evidently, the π-electron-rich 2-thiophenyl substituted indole 1m produced the acetoxylated compound 2m in low yield.
 |
| | Scheme 2 Scope of C-3 acetoxylation of N-heteroaryl substituted indoles. | |
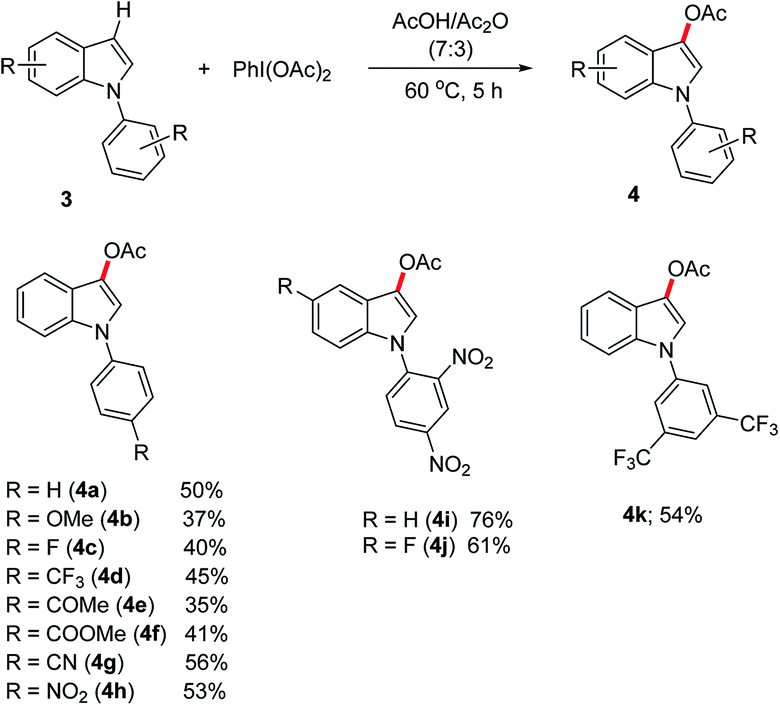
We further extended the metal-free acetoxylation method for the selective acetoxylation of the N-aryl substituted indoles. Hence, the different substituted N-aryl indoles 3 undergo acetoxylations at C-3 position to give the desired products in moderate yields (Scheme 3). Interestingly, a number of important functional groups like –F, –CF3, –C(O)Me, –C(O)OMe, –CN and –NO2 are well tolerated under the reaction conditions. To our surprise, the 2,4-dinitrophenyl substituted indoles 3i and 3j produced the acetoxylated products in good yields. The indoles containing N-aryl substituents with electron-withdrawing groups on the aryl backbone were found to produce improved yields of the C-3 acetoxylation, than the N-aryl substituents bearing electron-donating groups. Unlike free-NH-indole or indoles bearing electron-rich N-protecting groups, the N-aryl substituted indoles does not impart decomposition; however they produced 1-aryl-indolin-3-one and 2-oxo-1-aryl-indolin-3-yl acetate as the side products in different scale, which accounts for the low yields of the C-3 acetoxylated products.13 Most likely, these side products are formed from the hydrolysis of C-3 acetoxylated and diacetoxylated indoles.
 |
| | Scheme 3 Scope of C-3 acetoxylation of N-aryl substituted indoles. | |
In order to obtain the mechanistic insight, the intermolecular competition experiments between the indoles 1a and 3a as well as between 1a and 1m, were conducted (Scheme 4a and b); which highlighted that the indoles bearing π-electron-rich arene substituents were acetoxylated predominantly, though the yields were unsatisfactory. Further, an additional experiment between the different substituted N-pyrimidinyl-indoles 1a and 1c (Scheme 4c) clearly demonstrated that the electron-rich indole was preferentially acetoxylated, which revealed the nucleophilicity parameter14 of indoles for the acetoxylation process.
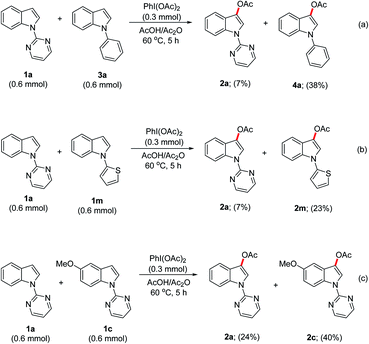 |
| | Scheme 4 Intermolecular competition experiments. | |
Further, to isolate the probable intermediate species, an experiment of 1a with PhI(OAc)2 was carried out at room temperature, which produced the diacetoxylated indoline 5a in 53% yield after 1 h (Scheme 5a). The formation of 5a might follow the pathway proposed for the similar compounds.12 Compound 5a led to the complete conversion into the acetoxylated product 2a upon heating in AcOH/Ac2O solvent for 3 h (Scheme 5b). This clearly demonstrated that the formation of 2a from 1a occurred via the intermediacy of 5a. The dehydroacetoxylation of 5a to 2a also occurred under non-acidic condition, however with low efficacy. Similar to the indoline 5a, the intermediate species for the indole 1m could not be observed; instead the direct formation of 2m was accomplished, including the intractable decomposed products.
 |
| | Scheme 5 Synthesis and reactivity of intermediate species. | |
To understand the dehydroacetoxylation process of the different N-substituted diacetoxyindolines, the compound 5a and 1-acetylindoline-2,3-diyl diacetate were heated in a single pot at 60 °C (Scheme 6). The compound 5a was completely converted into the C-3 acetoxylated product 2a in 2 h, whereas a trace amount of the C-3 acetoxylated product was formed from the 1-acetylindoline-2,3-diyl diacetate; suggesting that the dehydroacetoxylation of diacetoxylated indoline is significant in the presence of 2-pyrimidinyl as the N-substituent. Further, looking into the excellent reactivities of the indoles containing 2-Py, 2-Pym, 2-pyrazinyl or 2-nitroarene as the N-substituents (1a–1g, 1i–1l, 3i and 3j); we assume that the N-atom or N-group at the ortho-position of the substituents might have some additional influence on the dehydroacetoxylation reaction, in addition to the electronics of the substituents.
 |
| | Scheme 6 Dehydroacetoxylation of diacetoxylated compounds. | |
Conclusions
In summary, we have reported an efficient and regioselective method for the C-3 acetoxylation of the N-substituted indoles in the absence of metal-catalysts, wherein a broad range of functional groups are tolerated. The indoles containing π-electron-deficient arene substituents on the N-atom are acetoxylated efficiently than the one bearing strong sigma electron-donating or sigma electron-withdrawing substituents. The diacetoxylated indoline is proposed to be the active intermediate for the acetoxylation of the N-substituted indoles, where the dehydroacetoxylation is facilitated in the presence of a π-deficient arene substituents on the N-atom of indoles.
Experimental section
General information
All manipulations were conducted in an argon atmosphere using standard Schlenk techniques in pre-dried glassware. Liquid reagents were flushed with argon prior to use. The starting compounds, N-acyl-1H-indole,12 N-tosyl-1H-indole,12 N-benzyl-1H-indole,7 N-aryl-1H-indoles,15 N-pyridinyl-1H-indole,16 N-pyrimidinyl-1H-indole15a,17 and 1-acetylindoline-2,3-diyl diacetate12 were synthesized according to the previously described procedures. All other chemicals were obtained from commercial sources and were used without further purification. Representative starting compounds 1 and 3 as well as PhI(OAc)2 were analyzed by ICP-AES, which showed only trace amount of transition metals (<0.1 ppm Fe, Co, Ni, Cu, Pd, Rh and Ru). The yields refer to isolated compounds, estimated to be >95% pure as determined by 1H NMR. The 1H and 13C NMR spectra are referenced to the residual solvent signals (CDCl3: δ H = 7.26 ppm, δ C = 77.2 ppm).
Representative procedures for the preparation of N-substituted indoles
Representative procedure A. To a stirred solution of 1H-indole (0.120 g, 1.00 mmol) in DMF (15 mL) at 0 °C was added NaH (0.027 g, 1.10 mmol). After stirring the reaction mixture at 0 °C for 30 min, (hetero)aryl halide (1.20 mmol) was added and the reaction mixture was heated at 130 °C for 24 h. Then the reaction mixture was cooled to ambient temperature, poured into H2O (20 mL) and extracted with EtOAc (15 mL × 3). The combined organic phase was washed with H2O (15 mL × 3) and dried over Na2SO4. After filtration and evaporation of the solvents in vacuo, the crude product was purified by column chromatography on silica gel.
Representative procedure B. A mixture of 1H-indole (0.328 g, 2.80 mmol), (hetero)aryl halide (2.00 mmol), CuI (0.076 g, 0.40 mmol) and Cs2CO3 (1.30 g, 4.00 mmol) were taken in a flask and DMF (15 mL) was added into it. The reaction mixture was vigorously stirred at 120 °C under argon atmosphere for 40 h. Then the reaction mixture was cooled to ambient temperature, poured into H2O (20 mL) and extracted with EtOAc (15 mL × 3). The combined organic phase was washed with H2O (15 mL × 3) and dried over Na2SO4. After filtration and evaporation of the solvents in vacuo, the crude product was purified by column chromatography on silica gel.
2-Methyl-1-(pyrimidin-2-yl)-1H-indole (1f)18. The representative procedure A was followed using, 2-methyl-1H-indole (0.40 g, 3.05 mmol), 2-chloropyrimidine (0.489 g, 4.27 mmol), and NaH (0.095 g, 3.96 mmol). Purification by column chromatography on silica gel (hexane/EtOAc/Et3N: 20/1/0.5) yielded 1f (0.153 g, 24%) as a brown solid. M. p. = 48–50 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.79 (d, J = 4.9 Hz, 2H), 8.37 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 7.3 Hz, 1H), 7.31–7.24 (m, 2H), 7.11 (t, J = 4.9 Hz, 1H), 6.50 (s, 1H), 2.78 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 158.5, 158.1, 137.9, 137.0, 129.6, 122.5, 121.9, 119.6, 117.0, 114.1, 106.8, 16.7. IR (neat): νmax/cm−1 3032, 2962, 2853, 1562, 1417, 1305, 1205, 785, 733. HRMS (ESI) m/z calcd for C13H11N3 + H+ [M + H]+ 210.1026; found 210.1024.
1-(Pyrazin-2-yl)-1H-indole (1j)19. The representative procedure A was followed, using 1H-indole (0.120 g, 1.00 mmol), NaH (0.027 g, 1.10 mmol) and 2-iodopyrazine (0.246 g, 1.20 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 1j (0.186 g, 95%) as a brown solid. M. p. = 77–79 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.85 (s, 1H), 8.44 (d, J = 2.1 Hz, 1H), 8.36 (d, J = 2.1 Hz, 1H), 8.26 (d, J = 8.2 Hz, 1H), 7.68 (d, J = 3.4 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.31 (dd, J = 7.9, 7.3 Hz, 1H), 7.23 (dd, J = 7.9, 7.0 Hz, 1H), 6.74 (d, J = 3.4 Hz, 1H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 149.2, 142.8, 140.0, 136.4, 135.2, 130.7, 125.0, 123.9, 122.2, 121.4, 113.5, 107.3. IR (neat): νmax/cm−1 3108, 3053, 1450, 1360, 839, 739. HRMS (ESI) m/z calcd for C12H9N3 + H+ [M + H]+ 196.0869; found 196.0869.
5-Fluoro-1-(pyrazin-2-yl)-1H-indole (1k). The representative procedure A was followed, using 5-fluoro-1H-indole (0.23 g, 1.70 mmol), NaH (0.045 g, 1.87 mmol) and 2-iodopyrazine (0.420 g, 2.04 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 1k (0.33 g, 91%) as a brown solid. M. p. = 112–114 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.84 (d, J = 1.2 Hz, 1H), 8.47–8.46 (m, 1H), 8.41 (d, J = 2.7 Hz, 1H), 8.30 (dd, J = 9.1, 4.6 Hz, 1H), 7.72 (d, J = 3.7 Hz, 1H), 7.29 (dd, J = 9.1, 2.7 Hz, 1H), 7.06 (td, J = 9.1, 2.5 Hz, 1H), 6.71 (d, J = 3.4 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 159.0 (d, JC–F = 239.0 Hz), 149.1, 142.6, 140.2, 136.0, 131.8, 131.4 (d, JC–F = 10.0 Hz), 126.3, 114.9 (d, JC–F = 9.3 Hz), 111.9 (d, JC–F = 25.4 Hz), 107.2 (d, JC–F = 3.8 Hz), 106.5 (d, JC–F = 23.9 Hz). 19F-NMR (377 MHz, CDCl3): δ = −121.6 (s). IR (neat): νmax/cm−1 2921, 1469, 1443, 1353, 809, 753, 714, 616. HRMS (ESI) m/z calcd for C12H8FN3 + H+ [M + H]+ 214.0775; found 214.0775.
5-Bromo-1-(pyrazin-2-yl)-1H-indole (1l). The representative procedure A was followed, using 5-bromo-1H-indole (0.330 g, 1.70 mmol), NaH (0.045 g, 1.87 mmol) and 2-iodopyrazine (0.420 g, 2.04 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 1l (0.44 g, 95%) as off-white solid. M. p. = 98–100 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.84 (d, J = 1.2 Hz, 1H), 8.49–8.48 (m, 1H), 8.43 (d, J = 2.7 Hz, 1H), 8.20 (d, J = 8.8 Hz, 1H), 7.77 (d, J = 2.0 Hz, 1H), 7.69 (d, J = 3.4 Hz, 1H), 7.40 (dd, J = 8.8, 2.0 Hz, 1H), 6.69 (d, J = 3.2 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 149.0, 142.8, 140.5, 136.2, 134.0, 132.3, 126.7, 126.0, 123.9, 115.4, 115.3, 106.7. IR (neat): νmax/cm−1 2921, 1519, 1482, 1416, 1198, 1136, 1054, 1004, 957, 752, 709. HRMS (ESI) m/z calcd for C12H8BrN3 + H+ [M + H]+ 273.9974, 275.9954; found 273.9974, 275.9953.
1-(Thiophen-2-yl)-1H-indole (1m)15c. The representative procedure B was followed, using 1H-indole (0.328 g, 2.80 mmol), 2-bromothiophene (0.326 g, 2.00 mmol), CuI (0.076 g, 0.40 mmol) and Cs2CO3 (1.30 g, 4.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 100/1) yielded 1m (0.286 g, 72%) as a green liquid. 1H-NMR (400 MHz, CDCl3): δ = 7.64 (d, J = 7.8 Hz, 1H), 7.57 (d, J = 8.3 Hz, 1H), 7.27–7.22 (m, 2H), 7.17 (dd, J = 7.1, 6.9 Hz, 1H), 7.12 (dd, J = 5.6, 1.5 Hz, 1H), 7.03 (dd, J = 3.7, 1.5 Hz, 1H), 7.01–6.99 (m, 1H), 6.63 (d, J = 3.2 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 141.8, 137.2, 129.5, 129.2, 126.2, 123.0, 121.8, 121.2, 121.0, 120.6, 110.8, 104.3. IR (neat): νmax/cm−1 3108, 3009, 1611, 1550, 1459, 1312, 1226, 1201, 1013, 903, 842. HRMS (ESI) m/z calcd for C12H9NS + H+ [M + H]+ 200.0528; found 200.0526.
1-(4-(Trifluoromethyl)phenyl)-1H-indole (3d)20. The representative procedure B was followed, using 1H-indole (0.302 g, 2.58 mmol), 1-iodo-4-(trifluoromethyl)benzene (0.50 g, 1.84 mmol), CuI (0.070 g, 0.368 mmol) and Cs2CO3 (1.20 g, 3.68 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 20/1) yielded 3d (0.408 g, 85%) as a brown solid. M. p. = 50–52 °C. 1H-NMR (400 MHz, CDCl3): δ = 7.75 (d, J = 8.3 Hz, 2H), 7.68 (d, J = 7.6 Hz, 1H), 7.60–7.56 (m, 3H), 7.31 (d, J = 3.4 Hz, 1H), 7.24–7.17 (m, 2H), 6.70 (d, J = 3.2 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 143.0, 135.7, 129.9, 128.3 (q, JC–F = 32.4 Hz), 127.6, 127.1 (q, JC–F = 3.8 Hz), 124.2 (q, JC–F = 171.8 Hz), 124.1, 123.1, 121.6, 121.2, 110.5, 105.1. 19F-NMR (377 MHz, CDCl3): δ = −62.2 (s). IR (neat): νmax/cm−1 3058, 2962, 1607, 1521, 1455, 1318, 1158, 1061, 1013, 840. HRMS (ESI) m/z calcd for C15H10F3N + H+ [M + H]+ 262.0838; found 262.0837.
Methyl-4-(1H-indol-1-yl)benzoate (3f)15c. The representative procedure B was followed, using 1H-indole (0.328 g, 2.80 mmol), methyl 4-iodobenzoate (0.524 g, 2.00 mmol), CuI (0.076 g, 0.40 mmol) and K2CO3 (0.553 g, 4.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 10/1) yielded 3f (0.231 g, 46%) as a grey solid. M. p. = 55–57 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.18 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 7.9 Hz, 1H), 7.62 (d, J = 8.2 Hz, 1H), 7.58 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 3.4 Hz, 1H), 7.25 (t, J = 7.3 Hz, 1H), 7.19 (t, J = 7.3 Hz, 1H), 6.71 (d, J = 3.4 Hz, 1H), 3.95 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 166.6, 143.9, 135.6, 131.4, 130.0, 127.8, 127.6, 123.4, 123.0, 121.5, 121.1, 110.7, 105.1, 52.4. IR (neat): νmax/cm−1 3106, 3049, 2947, 1707, 1602, 1509, 1455, 1433, 1281, 1117, 1097, 720. HRMS (ESI) m/z calcd for C16H13NO2 + H+ [M + H]+ 252.1019; found 252.1012.
1-(4-Nitrophenyl)-1H-indole (3h)19,20. The representative procedure B was followed, using 1H-indole (0.328 g, 2.80 mmol), 1-iodo-4-nitrobenzene (0.50 g, 2.00 mmol), CuI (0.076 g, 0.40 mmol) and K2CO3 (0.553 g, 4.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 20/1) yielded 3h (0.224 g, 47%) as a yellow solid. M. p. = 132–134 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.33 (d, J = 8.9 Hz, 2H), 7.68 (d, J = 7.6 Hz, 1H), 7.62–7.60 (m, 3H), 7.33 (d, J = 3.1 Hz, 1H), 7.27 (dd, J = 7.6, 7.3 Hz, 1H), 7.21 (dd, J = 7.6, 7.3 Hz, 1H), 6.75 (d, J = 3.0 Hz, 1H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 145.3, 145.1, 135.4, 130.2, 127.2, 125.6, 123.5, 123.4, 121.8, 121.7, 110.6, 106.3. IR (neat): νmax/cm−1 3109, 1592, 1503, 1455, 1317, 1100, 882, 852, 690. HRMS (ESI) m/z calcd for C14H10N2O2 + H+ [M + H]+ 239.0815; found 239.0811.
1-(2,4-Dinitrophenyl)-1H-indole (3i)21. The representative procedure B was followed, using 1H-indole (0.351 g, 3.00 mmol), 1-chloro-2,4-dinitrobenzene (0.73 g, 3.60 mmol), CuI (0.114 g, 0.60 mmol), K2CO3 (0.830 g, 6.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 3i (0.424 g, 50%) as an orange solid. M. p. = 96–98 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.89 (d, J = 2.5 Hz, 1H), 8.54 (dd, J = 8.8, 2.5 Hz, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.71 (d, J = 6.6 Hz, 1H), 7.30–7.21 (m, 3H), 7.15 (d, J = 3.4 Hz, 1H), 6.83 (d, J = 3.2 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 145.4, 144.6, 138.0, 135.8, 129.7, 129.6, 128.1, 127.2, 123.9, 122.2, 121.9, 121.8, 109.4, 107.5. IR (neat): νmax/cm−1 3103, 1601, 1539, 1493, 1453, 1339, 1231, 1200, 844, 769, 752. HRMS (ESI) m/z calcd for C14H9N3O4 + H+ [M + H]+ 284.0666; found 284.0668.
1-(2,4-Dinitrophenyl)-5-fluoro-1H-indole (3j)22. The representative procedure B was followed, using 5-fluoro-1H-indole (0.203 g, 1.50 mmol), 1-chloro-2,4-dinitrobenzene (0.365 g, 1.80 mmol), CuI (0.057 g, 0.30 mmol), K2CO3 (0.415 g, 3.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 3j (0.138 g, 31%) as a yellow solid. M. p. = 151–153 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.90 (d, J = 2.2 Hz, 1H), 8.60 (dd, J = 8.8, 2.2 Hz, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.34 (dd, J = 9.1, 2.2 Hz, 1H), 7.17 (d, J = 3.4 Hz, 1H), 7.13–7.10 (m, 1H), 7.00 (td, J = 8.8, 2.2 Hz, 1H), 6.77 (d, J = 2.9 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 159.1 (d, JC–F = 238.1 Hz), 145.8, 145.0, 138.0, 132.6, 130.5 (d, JC–F = 10.0 Hz), 129.9, 128.9, 128.4, 122.0, 112.3 (d, JC–F = 27.0 Hz), 110.3 (d, JC–F = 9.2 Hz), 107.5 (d, JC–F = 4.6 Hz), 107.3 (d, JC–F = 23.9 Hz). 19F-NMR (377 MHz, CDCl3): δ = −121.5 (s). IR (neat): νmax/cm−1 3147, 3117, 3100, 1601, 1524, 1339, 1135, 908, 729. MS (EI) m/z: 301 [M]+, 281, 256, 226, 208, 181, 135.
1-(3,5-Bis(trifluoromethyl)phenyl)-1H-indole (3k)22. The representative procedure B was followed, using 1H-indole (0.328 g, 2.80 mmol), 1-bromo-3,5-bis(trifluoromethyl)benzene (0.586 g, 2.00 mmol), CuI (0.076 g, 0.40 mmol) and Cs2CO3 (1.30 g, 4.00 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 10/1) yielded 3k (0.222 g, 34%) as a light yellow liquid. 1H-NMR (400 MHz, CDCl3): δ = 7.97 (s, 2H), 7.84 (s, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 3.4 Hz, 1H), 7.29 (dd, J = 8.0, 7.3 Hz, 1H), 7.22 (dd, J = 7.6, 7.3 Hz, 1H), 6.75 (d, J = 3.2 Hz, 1H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 141.5, 135.6, 133.6 (q, JC–F = 34.0 Hz), 130.0, 127.3, 124.0 (q, JC–F = 3.0 Hz), 123.7, 123.1 (q, JC–F = 272.6 Hz), 121.9, 121.7, 119.8 (q, JC–F = 3.8 Hz), 109.9, 106.1. 19F-NMR (377 MHz, CDCl3): δ = −63.0 (s). IR (neat): νmax/cm−1 3284, 3062, 2963, 1477, 1402, 1275, 1115, 1103, 890, 847, 702. MS (EI) m/z: 329 [M]+, 310, 233, 213, 116.
Representative procedure for C-3 acetoxylation of N-substituted indoles
Synthesis of 1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2a). To a flame-dried Schlenk tube equipped with magnetic stir bar was introduced PhI(OAc)2 (0.106 g, 0.33 mmol) and 1-(pyrimidin-2-yl)-1H-indole (1a) (0.059 g, 0.30 mmol) under argon. The Schlenk tube with the mixture was evacuated and refilled with argon. To the above mixture was added acetic acid and acetic anhydride (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solvent mixture (1.0 mL). The resultant reaction mixture was then degassed, refilled with argon and stirred at 60 °C in a pre-heated oil bath for 5 h. At ambient temperature, H2O (5 mL) and saturated NaHCO3 solution (15 mL) were added and the reaction mixture was extracted with ethyl acetate (15 mL × 3). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo. The resultant residue was purified by column chromatography on silica gel (hexane/EtOAc: 5/1) to yielded acetoxylated compound 2a (0.071 g, 93%) as off-white solid. M. p. = 117–119 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.81 (d, J = 8.6 Hz, 1H), 8.66 (d, J = 4.7 Hz, 2H), 8.43 (s, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.25 (dd, J = 7.6, 7.3 Hz, 1H), 7.01 (t, J = 4.7 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.3, 158.3, 157.9, 133.7, 133.0, 124.8, 124.2, 122.3, 117.6, 116.6, 116.2, 114.7, 21.2. IR (neat): νmax/cm−1 3195, 2926, 1744, 1455, 1430, 1368, 1208, 809, 787. HRMS (ESI) m/z calcd for C14H11N3O2 + Na+ [M + Na]+ 276.0743; found 276.0738.
:3) solvent mixture (1.0 mL). The resultant reaction mixture was then degassed, refilled with argon and stirred at 60 °C in a pre-heated oil bath for 5 h. At ambient temperature, H2O (5 mL) and saturated NaHCO3 solution (15 mL) were added and the reaction mixture was extracted with ethyl acetate (15 mL × 3). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo. The resultant residue was purified by column chromatography on silica gel (hexane/EtOAc: 5/1) to yielded acetoxylated compound 2a (0.071 g, 93%) as off-white solid. M. p. = 117–119 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.81 (d, J = 8.6 Hz, 1H), 8.66 (d, J = 4.7 Hz, 2H), 8.43 (s, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.37 (t, J = 7.6 Hz, 1H), 7.25 (dd, J = 7.6, 7.3 Hz, 1H), 7.01 (t, J = 4.7 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.3, 158.3, 157.9, 133.7, 133.0, 124.8, 124.2, 122.3, 117.6, 116.6, 116.2, 114.7, 21.2. IR (neat): νmax/cm−1 3195, 2926, 1744, 1455, 1430, 1368, 1208, 809, 787. HRMS (ESI) m/z calcd for C14H11N3O2 + Na+ [M + Na]+ 276.0743; found 276.0738.
5-Methyl-1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2b). The representative procedure was followed, using 5-methyl-1-(pyrimidin-2-yl)-1H-indole (1b) (0.063 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 2b (0.061 g, 76%) as a brown solid. M. p. = 161–163 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.66 (d, J = 8.6 Hz, 1H), 8.62 (d, J = 4.9 Hz, 2H), 8.37 (s, 1H), 7.34 (s, 1H), 7.19 (d, J = 8.6, 1H), 6.95 (t, J = 4.7 Hz, 1H), 2.48 (s, 3H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.3, 158.1, 157.8, 133.4, 131.8, 131.3, 126.2, 124.3, 117.3, 116.3, 115.9, 114.7, 21.5, 21.1. IR (neat): νmax/cm−1 3202, 2917, 2851, 1747, 1579, 1451, 1430, 1206, 908, 785, 711. HRMS (ESI) m/z calcd for C15H13N3O2 + Na+ [M + Na]+ 290.0900; found 290.0894.
5-Methoxy-1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2c). The representative procedure was followed, using 5-methoxy-1-(pyrimidin-2-yl)-1H-indole (1c) (0.068 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 2c (0.064 g, 75%) as a light brown crystalline solid. M. p. = 166–168 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.69 (d, J = 9.5 Hz, 1H), 8.61 (d, J = 4.7 Hz, 2H), 8.39 (s, 1H), 6.99–6.95 (m, 3H), 3.89 (s, 3H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.2, 158.2, 157.7, 155.7, 133.5, 127.9, 124.8, 117.7, 116.0, 115.2, 114.1, 99.5, 55.8, 21.2. IR (neat): νmax/cm−1 3209, 2923, 1745, 1435, 1222, 1204, 911, 821, 776. HRMS (ESI) m/z calcd for C15H13N3O3 + Na+ [M + Na]+ 306.0849; found 306.0844.
5-Fluoro-1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2d). The representative procedure was followed, using 5-fluoro-1-(pyrimidin-2-yl)-1H-indole (1d) (0.064 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 3/1) yielded 2d (0.070 g, 86%) as an off-white solid. M. p. = 173–175 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.77 (dd, J = 9.2, 4.6 Hz, 1H), 8.66 (d, J = 4.9 Hz, 2H), 8.46 (s, 1H), 7.20 (dd, J = 6.7, 1.8 Hz, 1H), 7.09 (td, J = 9.2, 1.8 Hz, 1H), 7.03 (t, J = 4.9 Hz, 1H), 2.39 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.2, 159.1 (d, JC–F = 239.4 Hz), 158.3, 157.7, 133.4, 129.4, 124.9 (d, JC–F = 9.5 Hz), 117.9 (d, JC–F = 9.5 Hz), 116.4, 116.3, 112.7 (d, JC–F = 24.8 Hz), 103.2 (d, JC–F = 24.8 Hz), 21.1. 19F-NMR (377 MHz, CDCl3): δ = −121.0 (s). IR (neat): νmax/cm−1 3208, 2921, 2852, 1745, 1449, 1197, 792, 786, 588. HRMS (ESI) m/z calcd for C14H10FN3O2 + Na+ [M + Na]+ 294.0649; found 294.0647.
5-Bromo-1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2e). The representative procedure was followed, using 5-bromo-1-(pyrimidin-2-yl)-1H-indole (1e) (0.082 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 3/1) yielded 2e (0.073 g, 73%) as a light brown solid. M. p. = 178–179 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.68–8.65 (m, 3H), 8.42 (s, 1H), 7.67 (d, J = 2.0 Hz, 1H), 7.43 (dd, J = 9.2, 2.0 Hz, 1H), 7.04 (t, J = 4.9 Hz, 1H), 2.39 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.1, 158.3, 157.6, 132.6, 131.6, 127.6, 125.8, 120.3, 118.2, 116.5, 115.9, 115.7, 21.1. IR (neat): νmax/cm−1 3205, 2923, 1758, 1456, 1433, 1204, 956, 867, 784. HRMS (ESI) m/z calcd for C14H10BrN3O2 + Na+ [M + Na]+ 353.9849, 355.9828; found 353.9848, 355.9824.
2-Methyl-1-(pyrimidin-2-yl)-1H-indol-3-yl acetate (2f). The representative procedure was followed, using 2-methyl-1-(pyrimidin-2-yl)-1H-indole (1f) (0.075 g, 0.358 mmol) and PhI(OAc)2 (0.127 g, 0.394 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 2/1) yielded 2f (0.025 g, 26%) as a yellow solid. M. p. = 54–56 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.75 (d, J = 4.9 Hz, 2H), 8.42 (d, J = 8.2 Hz, 1H), 7.34 (d, J = 7.6, 1H), 7.27–7.19 (m, 2H), 7.11 (t, J = 4.9 Hz, 1H), 2.59 (s, 3H), 2.42 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 169.2, 158.5, 158.2, 134.1, 131.4, 127.0, 123.5, 123.1, 122.2, 117.1, 116.6, 114.8, 20.8, 12.8. IR (neat): νmax/cm−1 2923, 2852, 1756, 1575, 1559, 1421, 1366, 1205, 733. HRMS (ESI) m/z calcd for C15H13N3O2 + Na+ [M + Na+] 290.0900; found 290.0896.
1-(Pyridin-2-yl)-1H-indol-3-yl acetate (2g). The representative procedure was followed, using 1-(pyridin-2-yl)-1H-indole (1g) (0.058 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by preparative TLC (hexane/EtOAc: 5/1) and extraction in CH2Cl2 (15 mL × 3) yielded 2g (0.061 g, 81%) as a yellow liquid. 1H-NMR (500 MHz, CDCl3): δ = 8.54 (dd, J = 4.9, 1.2 Hz, 1H), 8.30 (d, J = 8.3 Hz, 1H), 7.95 (s, 1H), 7.79 (dd, J = 8.3, 7.3 Hz, 1H), 7.60 (d, J = 7.8 Hz, 1H), 7.44 (dd, J = 8.3, 0.5 Hz, 1H), 7.33 (ddd, J = 8.3, 7.1, 1.2 Hz, 1H), 7.24 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 7.14 (dd, J = 7.3, 4.9 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.4, 152.6, 149.0, 138.6, 133.0, 132.5, 124.3, 122.8, 121.5, 120.1, 117.8, 114.8, 114.4, 113.7, 21.2. IR (neat): νmax/cm−1 3178, 2962, 1752, 1449, 1435, 1357, 1207, 1009, 990, 799, 782. HR-MS (ESI) m/z calcd for C15H12N2O2 + H+ [M + H]+ 253.0972; found 253.0969.
1-(Pyridin-3-yl)-1H-indol-3-yl acetate (2h). The representative procedure was followed, using 1-(pyridin-3-yl)-1H-indole (1h) (0.058 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by preparative TLC (hexane/EtOAc: 5/1) and extraction in CH2Cl2 (15 mL × 3) yielded 2h (0.042 g, 56%) as a yellow liquid. 1H-NMR (500 MHz, CDCl3): δ = 8.84 (d, J = 1.8 Hz, 1H), 8.60 (d, J = 4.6 Hz, 1H), 7.83 (d, J = 7.9 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 7.59 (s, 1H), 7.50 (d, J = 8.2 Hz, 1H), 7.47 (dd, J = 7.9, 4.6 Hz, 1H), 7.28 (dd, J = 7.9, 7.3 Hz, 1H), 7.22 (dd, J = 7.6, 7.3 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.5, 147.5, 145.7, 136.3, 133.0, 132.5, 131.7, 124.4, 124.0, 121.8, 121.2, 118.3, 116.6, 110.2, 21.2. IR (neat): νmax/cm−1 3019, 3011, 1745, 1214, 746, 667. HRMS (ESI) m/z calcd for C15H12N2O2 + H+ [M + H]+ 253.0972; found 253.0969.
1-(Pyridin-4-yl)-1H-indol-3-yl acetate (2i). The representative procedure was followed, using 1-(pyridin-4-yl)-1H-indole (1i) (0.058 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by preparative TLC (hexane/EtOAc: 5/1) and extraction in CH2Cl2 (15 mL × 3) yielded 2i (0.042 g, 56%) as a yellow liquid. 1H-NMR (400 MHz, CDCl3): δ = 8.73 (d, J = 4.7 Hz, 2H), 7.75–7.66 (m, 3H), 7.49 (d, J = 4.9 Hz, 2H), 7.35 (dd, J = 7.8, 7.3 Hz, 1H), 7.29 (d, J = 7.3 Hz, 1H), 2.43 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.3, 151.4, 146.7, 133.4, 132.2, 124.5, 122.7, 121.7, 118.5, 117.4, 115.6, 111.0, 21.2. IR (neat): νmax/cm−1 2928, 2855, 1730, 1586, 1512, 1456, 1367, 1161, 993, 943, 807, 659. HRMS (ESI) m/z calcd for C15H12N2O2 + H+ [M + H]+ 253.0972; found 253.0971.
1-(Pyrazin-2-yl)-1H-indol-3-yl acetate (2j). The representative procedure was followed, using 1-(pyrazin-2-yl)-1H-indole (1j) (0.059 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by preparative TLC (hexane/EtOAc: 5/1) and extraction in CH2Cl2 (15 mL × 3) yielded 2j (0.071 g, 93%) as a brown solid. M. p. = 145–147 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.86 (d, J = 1.0 Hz, 1H), 8.48 (dd, J = 2.5, 1.7 Hz, 1H), 8.40 (d, J = 2.5 Hz, 1H), 8.34 (d, J = 8.6 Hz, 1H), 8.01 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.37 (dt, J = 8.1, 1.0 Hz, 1H), 7.28 (dd, J = 8.3, 7.8 Hz, 1H), 2.41 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.3, 149.3, 142.7, 140.0, 136.3, 134.1, 132.6, 125.0, 123.2, 122.3, 118.1, 114.0, 113.7, 21.2. IR (neat): νmax/cm−1 3021, 2961, 1737, 1449, 1425, 1365, 1210, 1123, 1013, 839, 729. HRMS (ESI) m/z calcd for C14H11N3O2 + Na+ [M + Na]+ 276.0743; found 276.0741.
5-Fluoro-1-(pyrazin-2-yl)-1H-indol-3-yl acetate (2k). The representative procedure was followed, using 5-fluoro-1-(pyrazin-2-yl)-1H-indole (1k) (0.064 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (CH2Cl2/EtOAc: 15/1) yielded 2k (0.050 g, 61%) as an off-white solid. M. p. = 166–168 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.81 (s, 1H), 8.47 (s, 1H), 8.41 (s, 1H), 8.37 (dd, J = 9.2, 4.3 Hz, 1H), 8.03 (s, 1H), 7.27–7.25 (m, 1H), 7.10 (td, J = 9.2, 2.4 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.1, 159.0 (d, JC–F = 240.3 Hz), 149.1, 142.6, 140.1, 135.8, 133.9, 129.1, 123.9 (d, JC–F = 9.5 Hz), 115.7 (d, JC–F = 9.5 Hz), 115.0, 113.2 (d, JC–F = 24.8 Hz), 103.5 (d, JC–F = 24.8 Hz), 21.2. 19F-NMR (377 MHz, CDCl3): δ = −121.6 (s). IR (neat): νmax/cm−1 2924, 1748, 1480, 1446, 1201, 1007, 911, 839, 764. HRMS (ESI) m/z calcd for C14H10FN3O2 + H+ [M + H]+ 272.0830; found 272.0828.
5-Bromo-1-(pyrazin-2-yl)-1H-indol-3-yl acetate (2l). The representative procedure was followed, using 5-bromo-1-(pyrazin-2-yl)-1H-indole (1l) (0.082 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (CH2Cl2/EtOAc: 20/1) yielded 2l (0.069 g, 70%) as a light brown solid. M. p. = 162–164 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.81 (s, 1H), 8.48 (s, 1H), 8.42 (d, J = 2.5 Hz, 1H), 8.27 (d, J = 9.1 Hz, 1H), 8.00 (s, 1H), 7.75 (d, J = 1.7 Hz, 1H), 7.44 (dd, J = 8.8, 1.7 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.1, 149.0, 142.6, 140.4, 136.0, 133.2, 131.2, 127.9, 124.8, 120.8, 115.9, 115.6, 114.6, 21.2. IR (neat): νmax/cm−1 3170, 2923, 1759, 1446, 1422, 1243, 1061, 1006, 835, 735. HRMS (ESI) m/z calcd for C14H10BrN3O2 + H+ [M + H]+ 332.0029, 334.0009; found 332.0030, 334.0008.
1-(Thiophen-2-yl)-1H-indol-3-yl acetate (2m). The representative procedure was followed, using 1-(thiophen-2-yl)-1H-indole (1m) (0.043 g, 0.22 mmol) and PhI(OAc)2 (0.077 g, 0.24 mmol). Purification by preparative TLC (hexane/EtOAc: 5/1) and extraction in CH2Cl2 (15 mL × 3) yielded 2m (0.020 g, 35%) as a liquid. 1H-NMR (400 MHz, CDCl3): δ = 7.60–7.54 (m, 3H), 7.29–7.25 (m, 1H), 7.22–7.16 (m, 2H), 7.08–7.02 (m, 2H), 2.38 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.4, 141.4, 134.4, 131.9, 126.2, 124.0, 122.0, 121.4, 121.1, 120.9, 118.5, 118.0, 110.9, 21.2. IR (neat): νmax/cm−1 3010, 2962, 2854, 1747, 1613, 1547, 1460, 1366, 1225, 1010, 692. HRMS (ESI) m/z calcd for C14H11NO2S + Na+ [M + Na]+ 280.0403; found 280.0398.
1-Phenyl-1H-indol-3-yl acetate (4a)7. The representative procedure was followed, using 1-phenyl-1H-indole (3a) (0.058 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 4a (0.038 g, 50%) as a liquid. 1H-NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 7.6 Hz, 1H), 7.57 (s, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.51–7.48 (m, 4H), 7.35–7.31 (m, 1H), 7.23 (dd, J = 7.6, 7.3 Hz, 1H), 7.18 (dd, J = 7.6, 7.3 Hz, 1H), 2.38 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.6, 139.6, 133.0, 131.7, 129.8, 126.6, 124.6, 123.4, 121.4, 120.6, 118.0, 117.2, 110.8, 21.2. IR (neat): νmax/cm−1 3007, 2978, 2873, 1745, 1712, 1501, 1457, 1367, 1223, 1110, 696. HRMS (ESI) m/z calcd for C16H13NO2 + Na+ [M + Na]+ 274.0838; found 274.0833.
1-(4-Methoxyphenyl)-1H-indol-3-yl acetate (4b)7. The representative procedure was followed, using 1-(4-methoxyphenyl)-1H-indole (3b) (0.067 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 4/1) yielded 4b (0.031 g, 37%) as a liquid. 1H-NMR (400 MHz, CDCl3): δ = 7.60 (d, J = 7.6 Hz, 1H), 7.49 (s, 1H), 7.43–7.37 (m, 3H), 7.24–7.14 (m, 2H), 7.01 (d, J = 8.6 Hz, 2H), 3.86 (s, 3H), 2.38 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.7, 158.5, 133.5, 132.5, 131.2, 126.2, 123.2, 120.9, 120.3, 117.9, 117.6, 114.9, 110.6, 55.8, 21.2. IR (neat): νmax/cm−1 3176, 2966, 2840, 1747, 1548, 1510, 1370, 1207, 1029, 739, 588. HRMS (ESI) m/z calcd for C17H15NO3 + Na+ [M + Na]+ 304.0944; found 304.0935.
1-(4-Fluorophenyl)-1H-indol-3-yl acetate (4c). The representative procedure was followed, using 1-(4-fluorophenyl)-1H-indole (3c) (0.076 g, 0.36 mmol) and PhI(OAc)2 (0.127 g, 0.39 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 15/1) yielded 4c (0.039 g, 40%) as a yellow liquid. 1H-NMR (400 MHz, CDCl3): δ = 7.67 (d, J = 7.6 Hz, 1H), 7.56 (s, 1H), 7.49–7.46 (m, 3H), 7.30–7.21 (m, 4H), 2.43 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.6, 161.2 (d, JC–F = 246.6 Hz), 135.6 (d, JC–F = 2.3 Hz), 133.3, 131.7, 126.4 (d, JC–F = 8.5 Hz), 123.5, 121.3, 120.6, 118.0, 117.2, 116.6 (d, JC–F = 23.1 Hz), 110.4, 21.2. 19F-NMR (377 MHz, CDCl3): δ = −115.1 (s). IR (neat): νmax/cm−1 3160, 2927, 2854, 1745, 1509, 1366, 1202, 1128, 819, 738, 558. HRMS (ESI) m/z calcd for C16H12FNO2 + Na+ [M + Na]+ 292.0744; found 292.0739.
1-(4-(Trifluoromethyl)phenyl)-1H-indol-3-yl acetate (4d). The representative procedure was followed, using 1-(4-(trifluoromethyl)phenyl)-1H-indole (3d) (0.078 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 10/1) yielded 4d (0.043 g, 45%) as a brown solid. M. p. = 99–101 °C. 1H-NMR (400 MHz, CDCl3): δ = 7.79–7.76 (m, 2H), 7.66–7.61 (m, 4H), 7.58 (dd, J = 8.3, 4.9 Hz, 1H), 7.30–7.20 (m, 2H), 2.40 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.5, 142.7, 132.8, 132.6, 128.4 (q, JC–F = 33.1 Hz), 127.1 (q, JC–F = 3.1 Hz), 124.2 (q, JC–F = 272.0 Hz), 124.1, 124.0, 122.0, 121.2, 118.3, 116.6, 110.6, 21.2. 19F-NMR (377 MHz, CDCl3): δ = −62.3 (s). IR (neat): νmax/cm−1 3163, 2926, 1746, 1369, 1333, 1106, 1065, 841, 763, 736. HRMS (ESI) m/z calcd for C17H12F3NO2 + Na+ [M + Na]+ 342.0712; found 342.0706.
1-(4-Acetylphenyl)-1H-indol-3-yl acetate (4e). The representative procedure was followed, using 1-(4-(1H-indol-1-yl)phenyl)ethan-1-one (3e) (0.071 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 3/1) yielded 4e (0.031 g, 35%) as a light yellow liquid. 1H-NMR (500 MHz, CDCl3): δ = 8.10 (d, J = 8.5 Hz, 2H), 7.64–7.58 (m, 5H), 7.28 (dd, J = 8.2, 7.0 Hz, 1H), 7.22 (dd, J = 7.6, 7.3 Hz, 1H), 2.64 (s, 3H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 197.0, 168.5, 143.6, 134.7, 132.7, 132.6, 130.2, 124.0, 123.5, 122.1, 121.3, 118.3, 116.5, 110.8, 26.8, 21.2. IR (neat): νmax/cm−1 2962, 2931, 2861, 1717, 1265, 1128, 1074, 736. HRMS (ESI) m/z calcd for C18H15NO3 + Na+ [M + Na]+ 316.0944; found 316.0938.
Methyl 4-(3-acetoxy-1H-indol-1-yl)benzoate (4f). The representative procedure was followed, using methyl 4-(1H-indol-1-yl)benzoate (3f) (0.075 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 4f (0.038 g, 41%) as a liquid. 1H-NMR (500 MHz, CDCl3): δ = 8.18 (d, J = 8.5 Hz, 2H), 7.64–7.60 (m, 3H), 7.57 (d, J = 8.5 Hz, 2H), 7.28 (dd, J = 8.2, 7.3 Hz, 1H), 7.22 (dd, J = 7.6, 7.3 Hz, 1H), 3.96 (s, 3H), 2.40 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.5, 166.5, 143.5, 132.7, 132.6, 131.4, 127.7, 124.0, 123.4, 122.0, 121.2, 118.2, 116.5, 110.8, 52.4, 21.2. IR (neat): νmax/cm−1 3028, 3012, 2954, 1717, 1605, 1515, 1457, 1437, 1365, 1280, 1206, 738, 664. HRMS (ESI) m/z calcd for C18H15NO4 + Na+ [M + Na]+ 332.0893; found 332.0887.
1-(4-Cynophenyl)-1H-indol-3-yl acetate (4g). The representative procedure was followed, using 4-(1H-indol-1-yl)benzonitrile (3g) (0.066 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 4g (0.046 g, 56%) as a light brown solid. M. p. = 110–112 °C. 1H-NMR (500 MHz, CDCl3): δ = 7.81 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 7.6 Hz, 1H), 7.66 (s, 1H), 7.63–7.60 (m, 3H), 7.33 (dd, J = 8.2, 7.3 Hz, 1H), 7.27 (dd, J = 8.2, 7.0 Hz, 1H), 2.43 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.4, 143.4, 133.9, 133.1, 132.4, 124.3, 124.0, 122.3, 121.6, 118.5, 118.4, 116.1, 110.6, 109.4, 21.2. IR (neat): νmax/cm−1 2921, 2851, 2225, 1776, 1562, 1509, 1366, 1211, 1126, 1012, 845, 743. HRMS (ESI) m/z calcd for C17H12N2O2 + Na+ [M + Na]+ 299.0791; found 299.0787.
1-(4-Nitrophenyl)-1H-indol-3-yl acetate (4h). The representative procedure was followed, using 1-(4-nitrophenyl)-1H-indole (3h) (0.071 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 5/1) yielded 4h (0.047 g, 53%) as a yellow solid. M. p. = 142–144 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.38 (d, J = 8.9 Hz, 2H), 7.68–7.62 (m, 5H), 7.33 (dd, J = 8.2, 7.3 Hz, 1H), 7.24 (dd, J = 7.6, 7.3 Hz, 1H), 2.41 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.4, 145.2, 145.1, 133.5, 132.5, 125.7, 124.6, 123.5, 122.5, 121.8, 118.6, 116.2, 110.7, 21.2. IR (neat): νmax/cm−1 2923, 2852, 1746, 1592, 1524, 1222, 1129, 865, 748, 695, 528. HRMS (ESI) m/z calcd for C16H12N2O4 + Na+ [M + Na]+ 319.0689; found 319.0688.
1-(2,4-Dinitrophenyl)-1H-indol-3-yl acetate (4i). The representative procedure was followed, using 1-(2,4-dinitrophenyl)-1H-indole (3i) (0.085 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 4:1) yielded 4i (0.078 g, 76%) as an orange solid. M. p. = 147–148 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.92 (d, J = 2.4 Hz, 1H), 8.57 (dd, J = 8.9, 2.4 Hz, 1H), 7.84 (d, J = 8.9 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.53 (s, 1H), 7.33–7.27 (m, 2H), 7.16 (d, J = 8.2 Hz, 1H), 2.42 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 168.0, 145.5, 144.7, 138.0, 134.3, 133.1, 130.0, 128.3, 125.0, 122.3, 122.2, 122.1, 118.8, 116.2, 109.5, 21.2. IR (neat): νmax/cm−1 3158, 3084, 2852, 1751, 1601, 1527, 1366, 1224, 1128, 748, 702, 569. HRMS (ESI) m/z calcd for C16H11N3O6 + Na+ [M + Na]+ 364.0540; found 364.0533.
1-(2,4-Dinitrophenyl)-5-fluoro-1H-indol-3-yl acetate (4j). The representative procedure was followed, using 1-(2,4-dinitrophenyl)-5-fluoro-1H-indole (3j) (0.09 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 4:1) yielded 4j (0.066 g, 61%) as an orange solid. M. p. = 120–122 °C. 1H-NMR (400 MHz, CDCl3): δ = 8.90 (d, J = 2.4 Hz, 1H), 8.57 (dd, J = 8.8, 2.4 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.55 (s, 1H), 7.30 (dd, J = 8.6, 2.2 Hz, 1H), 7.08–6.99 (m, 2H), 2.39 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 167.9, 159.0 (d, JC–F = 239.7 Hz), 145.8, 144.9, 137.7, 134.0 (d, JC–F = 4.6 Hz), 130.1, 129.6, 128.4, 122.9 (d, JC–F = 10.8 Hz), 122.1, 117.9, 113.5 (d, JC–F = 26.2 Hz), 110.7 (d, JC–F = 9.3 Hz), 104.4 (d, JC–F = 25.4 Hz), 21.1. 19F-NMR (377 MHz, CDCl3): δ = −120.4 (s). IR (neat): νmax/cm−1 2922, 2852, 1754, 1452, 1338, 1202, 1188, 769, 729, 521. HRMS (ESI) m/z calcd for C16H10FN3O6 + Na+ [M + Na]+ 382.0446; found 382.0445.
1-(3,5-Bis(trifluoromethyl)phenyl)-1H-indol-3-yl acetate (4k). The representative procedure was followed, using 1-(3,5-bis(trifluoromethyl)phenyl)-1H-indole (3k) (0.099 g, 0.30 mmol) and PhI(OAc)2 (0.106 g, 0.33 mmol). Purification by column chromatography on silica gel (hexane/EtOAc: 20/1) yielded 4k (0.063 g, 54%) as a light brown solid. M. p. = 68–70 °C. 1H-NMR (400 MHz, CDCl3): δ = 7.96 (s, 2H), 7.83 (s, 1H), 7.66–7.64 (m, 2H), 7.50 (d, J = 8.3 Hz, 1H), 7.32 (dd, J = 7.3, 7.1 Hz, 1H), 7.24 (dd, J = 7.6, 7.3 Hz, 1H), 2.40 (s, 3H). 13C{1H}-NMR (100 MHz, CDCl3): δ = 168.4, 141.1, 133.5 (q, J = 33.3 Hz), 133.2, 132.7, 124.6, 124.0 (q, J = 2.9 Hz), 123.1 (q, J = 272.8 Hz), 122.2, 121.7, 119.7, 118.6, 116.2, 110.0, 21.1. 19F-NMR (377 MHz, CDCl3): δ = −63.0 (s). IR (neat): νmax/cm−1 3185, 3066, 2927, 1750, 1279, 1216, 1202, 1168, 1085, 888, 737, 682. HRMS (ESI) m/z calcd for C18H11F6NO2 + H+ [M + H]+ 388.0767; found 388.0762.
Synthesis of 1-(pyrimidin-2-yl)indoline-2,3-diyl diacetate (5a). To a flame-dried Schlenk tube equipped with magnetic stir bar was introduced phenyl-λ3-iodanediyl diacetate, PhI(OAc)2 (0.354 g, 1.1 mmol) and 1-(pyrimidin-2-yl)-1H-indole (1a) (0.195 g, 1.0 mmol) under argon. The Schlenk tube with reaction mixture was evacuated and refilled with argon. To the above mixture was added acetic acid and acetic anhydride (7:3) solvent mixture (1.0 mL). The resultant reaction mixture was then degassed, refilled with argon and stirred at room temperature for 1 h. Then the reaction mixture was quenched with H2O (5 mL) and saturated NaHCO3 solution (15 mL) and the reaction mixture was extracted with ethyl acetate (15 mL × 3). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo. The crude residue was purified by column chromatography on silica gel (hexane/EtOAc: 2/1) to yielded diacyloxylated compound 5a (0.165 g, 53%) as an off-white solid. M. p. = 143–145 °C. 1H-NMR (500 MHz, CDCl3): δ = 8.54 (d, J = 4.6 Hz, 2H), 8.45 (d, J = 8.2 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.43 (dd, J = 8.2, 7.3 Hz, 1H), 7.22 (s, 1H), 7.06 (dd, J = 7.6, 7.3 Hz, 1H), 6.85 (t, J = 4.6 Hz, 1H), 6.01 (s, 1H), 2.07 (s, 3H), 2.05 (s, 3H). 13C{1H}-NMR (125 MHz, CDCl3): δ = 170.2, 170.1, 158.4, 158.0, 144.4, 131.0, 127.6, 127.4, 122.9, 116.2, 114.0, 87.9, 75.6, 21.1. IR (neat): νmax/cm−1 3735, 3015, 1734, 1730, 1585, 1488, 1444, 1227, 1014, 761, 617. HRMS (ESI) m/z calcd for C16H15N3O4 + Na+ [M + Na]+ 336.0955; found 336.0952.
Representative procedure for the competition experiments
To a flame-dried Schlenk tube equipped with magnetic stir bar was introduced compound 1a (0.117 g, 0.6 mmol), compound 3a (0.116 g, 0.6 mmol) and PhI(OAc)2 (0.097 g, 0.30 mmol) under argon. The Schlenk tube with the mixture was evacuated and refilled with argon. To the above mixture was added acetic acid and acetic anhydride (7:3) solvent mixture (2.0 mL). The resultant reaction mixture was then degassed, refilled with argon and stirred at 60 °C in a pre-heated oil bath for 5 h. At ambient temperature, H2O (5 mL) and saturated NaHCO3 solution (15 mL) were added and the reaction mixture was extracted with ethyl acetate (15 mL × 3). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo. To the resultant residue, acetone (5 mL) and p-xylene (0.3 mmol) were added and injected into Gas Chromatograph. The GC yield was calculated with respect to internal standard p-xylene.
Procedure for the dehydroacetoxylation reaction
To a flame-dried Schlenk tube equipped with magnetic stir bar was introduced compound 5a (0.031 g, 0.1 mmol) and 1-acetylindoline-2,3-diyl diacetate (0.028 g, 0.1 mmol). To the above mixture was added acetic acid and acetic anhydride (7:3) solvent mixture (1.0 mL). The resultant reaction mixture was then degassed, refilled with argon and stirred at 60 °C in a pre-heated oil bath for 2 h. At ambient temperature, H2O (3 mL) and saturated NaHCO3 solution (10 mL) were added and the reaction mixture was extracted with ethyl acetate (10 mL × 3). The combined organic layers were dried over Na2SO4 and the solvent was evaporated in vacuo. To the resultant residue, acetone (5 mL) and p-xylene (0.2 mmol) were added and injected into Gas Chromatograph. The GC yield was calculated with respect to internal standard p-xylene.
Acknowledgements
This work was financially supported by CSIR-NCL (start-up grant, MLP025926), SERB, New Delhi (SR/S1/IC-42/2012) and CSIR, New Delhi as part of XII Five Year plan programme under title ORIGIN (CSC-0108). V.S. and U.N.P. thank CSIR, New Delhi for research fellowship. We thank Dr P. R. Rajmohanan for NMR facility, Dr (Mrs) Shanthakumari for HRMS and Dr C. V. V. Satyanarayana for ICP-AES analyses.
Notes and references
-
(a) D. A. Alonso, C. Nájera, I. M. Pastor and M. Yus, Chem.–Eur. J., 2010, 16, 5274 CrossRef CAS PubMed;
(b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed;
(c) B. Liu and B.-F. Shi, Tetrahedron Lett., 2015, 56, 15 CrossRef CAS PubMed.
-
(a) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070 CrossRef CAS PubMed;
(b) V. S. Thirunavukkarasu and L. Ackermann, Org. Lett., 2012, 14, 6206 CrossRef CAS PubMed;
(c) V. S. Thirunavukkarasu, J. Hubrich and L. Ackermann, Org. Lett., 2012, 14, 4210 CrossRef CAS PubMed;
(d) Y. Yang, Y. Lin and Y. Rao, Org. Lett., 2012, 14, 2874 CrossRef CAS PubMed;
(e) X. Yang, G. Shan and Y. Rao, Org. Lett., 2013, 15, 2334 CrossRef CAS PubMed;
(f) F. Yang and L. Ackermann, Org. Lett., 2013, 15, 718 CrossRef CAS PubMed;
(g) G. Shan, X. Han, Y. Lin, S. Yu and Y. Rao, Org. Biomol. Chem., 2013, 11, 2318 RSC;
(h) F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285 CrossRef CAS PubMed;
(i) X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed.
-
(a) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed;
(b) T.-S. Jiang and G.-W. Wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed;
(c) W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362 CrossRef CAS PubMed;
(d) F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC;
(e) L.-B. Zhang, X.-Q. Hao, S.-K. Zhang, Z.-J. Liu, X.-X. Zheng, J.-F. Gong, J.-L. Niu and M.-P. Song, Angew. Chem., Int. Ed., 2015, 54, 272 CrossRef CAS PubMed.
-
(a) L. Eberson and L. Jonsson, J. Chem. Soc., Chem. Commun., 1974, 885 RSC;
(b) T. Yoneyama and R. H. Crabtree, J. Mol. Catal. A: Chem., 1996, 108, 35 CrossRef CAS;
(c) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed;
(d) W. Wang, F. Luo, S. Zhang and J. Cheng, J. Org. Chem., 2010, 75, 2415 CrossRef CAS PubMed;
(e) M. H. Emmert, A. K. Cook, Y. J. Xie and M. S. Sanford, Angew. Chem., Int. Ed., 2011, 50, 9409 CrossRef CAS PubMed;
(f) A. Pradal, P. F. dit Bel, P. Y. Toullec and V. Michelet, Synthesis, 2012, 44, 2463 CrossRef CAS PubMed;
(g) A. V. Gulevich, F. S. Melkonyan, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2012, 134, 5528 CrossRef CAS PubMed;
(h) M. R. Yadav, R. K. Rit and A. K. Sahoo, Chem.–Eur. J., 2012, 18, 5541 CrossRef CAS PubMed;
(i) J. B. Gary, A. K. Cook and M. S. Sanford, ACS Catal., 2013, 3, 700 CrossRef CAS;
(j) H. Zhang, R.-B. Hu, X.-Y. Zhang, S.-X. Li and S.-D. Yang, Chem. Commun., 2014, 50, 4686 RSC;
(k) R. K. Rit, M. R. Yadav and A. K. Sahoo, Org. Lett., 2014, 16, 968 CrossRef CAS PubMed;
(l) K. Chen, S.-Q. Zhang, H.-Z. Jiang, J.-W. Xu and B.-F. Shi, Chem.–Eur. J., 2015, 21, 3264 CrossRef CAS PubMed;
(m) S. Zhao, F.-J. Chen, B. Liu and B.-F. Shi, Sci. China: Chem., 2015 DOI:10.1007/s11426-015-5376-z.
-
(a) H. Zaimoku, T. Hatta, T. Taniguchi and H. Ishibashi, Org. Lett., 2012, 14, 6088 CrossRef CAS PubMed;
(b) Y. Chi, W.-X. Zhang and Z. Xi, Org. Lett., 2014, 16, 6274 CrossRef CAS PubMed.
- R. D. Arnold, W. M. Nutter and W. L. Stepp, J. Org. Chem., 1959, 24, 117 CrossRef CAS.
- P. Y. Choy, C. P. Lau and F. Y. Kwong, J. Org. Chem., 2011, 76, 80 CrossRef CAS PubMed For synthesis of 5-HT6 receptor mimics, see: K. Alex, N. Schwarz, V. Khedkar, I. A. Sayyed, A. Tillack, D. Michalik, J. Holenz, J. L. Diaz and M. Beller, Org. Biomol. Chem., 2008, 6, 1802 Search PubMed.
- Q. Liu, G. Li, H. Yi, P. Wu, J. Liu and A. Lei, Chem.–Eur. J., 2011, 17, 2353 CrossRef CAS PubMed.
-
(a) I. Mutule, E. Suna, K. Olofsson and B. Pelcman, J. Org. Chem., 2009, 74, 7195 CrossRef CAS PubMed;
(b) D. Lubriks, I. Sokolovs and E. Suna, Org. Lett., 2011, 13, 4324 CrossRef CAS PubMed.
-
(a) V. P. Mehta and B. Punji, RSC Adv., 2013, 3, 11957 RSC;
(b) C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS PubMed.
- K. Liu, P. Wen, J. Liu and G. Huang, Synthesis, 2010, 3623 CAS.
- Q. Liu, Q. Y. Zhao, J. Liu, P. Wu, H. Yi and A. Lei, Chem. Commun., 2012, 48, 3239 RSC.
- The 1-aryl-indolin-3-one (A) and 2-oxo-1-aryl-indolin-3-yl acetate (B) were isolated as mixture. Their authenticities have been proved from the 1H, 13C and GC-MS spectra of (A + B) mixture
. -
(a) S. Lakhdar, M. Westermaier, F. Terrier, R. Goumont, T. Boubaker, A. R. Ofial and H. Mayr, J. Org. Chem., 2006, 71, 9088 CrossRef CAS PubMed;
(b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66 CrossRef CAS PubMed.
-
(a) S. Xu, X. Huang, X. Hong and B. Xu, Org. Lett., 2012, 14, 4614 CrossRef CAS PubMed;
(b) Z.-L. Xu, H.-X. Li, Z.-G. Ren, W.-Y. Du, W.-C. Xu and J.-P. Lang, Tetrahedron, 2011, 67, 5282 CrossRef CAS PubMed;
(c) R. Xiao, H. Zhao and M. Cai, Tetrahedron, 2013, 69, 5444 CrossRef CAS PubMed.
-
(a) L. Ackermann and A. V. Lygin, Org. Lett., 2011, 13, 3332 CrossRef CAS PubMed;
(b) Y.-C. Teo, F.-F. Yong and S. Sim, Tetrahedron, 2013, 69, 7279 CrossRef CAS PubMed.
- J. Shi, B. Zhou, Y. Yang and Y. Li, Org. Biomol. Chem., 2012, 10, 8953 CAS.
- D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 2754 CrossRef CAS PubMed.
- J.-P. Wan, Y.-F. Chai, J.-M. Wu and Y.-J. Pan, Synlett, 2008, 3068 CAS.
- F. Bellina, C. Calandri, S. Cauteruccio and R. Rossi, Eur. J. Org. Chem., 2007, 2147 CrossRef CAS PubMed.
- R. Chicharro, S. de Castro, J. L. Reino and V. J. Arán, Eur. J. Org. Chem., 2003, 2314 CrossRef CAS PubMed.
- A satisfactory HRMS data was not observed for this compound.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10428a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.