
A novel phenylacetylene-indolium fluorophore for detection of cyanide by the naked eye†
Nakorn Niamnont*a,
Apiwat Promchatb,
Chutima Siangmaa,
Chuenjai Pramaulpornsatita and
Mongkol Sukwattanasinittb
aDepartment of Chemistry, Faculty of Science, King Mongkut's University of Technology Thonburi, Bangkok 10140, Thailand. E-mail: nakorn.nia@kmutt.ac.th; Tel: +66 89 183 7284
bDepartment of Chemistry, Faculty of Science and Nanotec-CU Center of Excellence on Food and Agriculture Chulalongkorn University, Bangkok 10330, Thailand
First published on 23rd July 2015
Abstract
A novel fluorescent turn-on cyanide sensor containing an indolium salt as the selective CN− receptor was synthesized in 2 steps via Sonogashira coupling and Knoevenagel reaction. Compound 2 was developed as a new fluorometric and colorimetric sensor for cyanide with a sensing mechanism through cyanide ion addition to the indolium group. Importantly, the inclusion complexes between compound 2 and α-cyclodrextin, exhibited fluorescent turn-on detection of CN− in 100% aqueous media with the detection limit of 1.3 μM. The turn-on signal is the result of the conversion of the conjugated hemicyanine moiety to a cyano-hemicyanine group via the addition of the cyanide ion. Using paper sensor strips, naked eye detection of cyanide is possible down to 4.59 ng mm−2. Test strips based on compound 2 can act as a convenient and efficient CN− testing kit.
Introduction
The cyanide anion (CN−) is one of the most poisonous chemicals to the health of humans and animals.1 The World Health Organization (WHO) designates the maximum level of cyanide permissible in drinking water as 1.9 μM.2 On the other hand, cyanide has been used in many industrial processes such as the plastics industry, metal mining, and electroplating processes.3 Additionally, cyanide is produced in nature by certain algae and the cassava plant.4 Cyanide contamination in wastewater, ground water, river water, and drinking water has been seriously recognized.5To detect cyanide anions, fluorescent and/or colorimetric sensors are highly attractive for their rapid detection and ease of use for on-site analyses where instrument-free methods are usually desirable.6 For organic compounds, a few numbers of functional groups such as boronic acid,7 aldehyde,8 and dicyanovinyl9 have been reported as selective CN− receptors. However, the sensors constructed from these functional groups have cross reactivity with other anions such as fluoride, acetate and phosphate ions and usually require some long response time to give stable signals.10
Recently, indolium has emerged as a very promising probe for CN− because it provided fast response and dual modes of detection in both fluorescence and color change.11 However, most of these cyanide sensors have been operated in solutions containing significant amount of organic solvents. As common cyanide salts are soluble in water and real samples are more common in aqueous media, it is thus of interest to have a cyanide sensor operate well in medium containing mostly water. Paper based indicator for naked eye detection of cyanide ion can also be very handy for on-site analyses.
Due to their strong fluorescence and good stability, we have recently utilized arylene-ethynylene as fluorogenic building blocks for metal ion, proteins and DNA sensors.12 We also found that water solubility of the fluorophore containing rod-shaped phenylene-ethynylene moieties could be significantly enhanced via the formation of inclusion complexes with cyclodextrins (CyD's).13 In this work, a novel cyanide fluorescent turn-on sensing compound 2 was designed based on phenylene-ethynylene fluorogenic rod attached to indolium group serving as a CN− receptor (Scheme 1). Compound 2 should also be able to form an inclusion complex with CyD's that lead to its application for cyanide detection in aqueous media.
 | ||
| Scheme 1 The synthetic routes of the compound 2. | ||
Experimental
Reagents and apparatus
Benzyltrimethylammonium chloride, potassium carbonate, and were purchased from Fluka. Triphenylamine, iodine monochloride, diisopropylamine, copper(I) iodide, and, bis(triphenylphosphine)palladium(II) dichloride, indolium salts, N,N-dimethylaniline, potassium hydroxide, triphenylphosphine, trimethylsilylethyne, and sodium thiosulfate were purchased from Aldrich. All other reagents were non-selectively purchased from Sigma-Aldrich, Fluka or Merck and used without further purification. Generally, solvents such as dichloromethane and acetonitrile were reagent grade stored over 4A molecular sieves. However, for anhydrous reactions, solvents such as THF and toluene were dried and distilled before use according to the standard procedures. All column chromatography was operated using Merck silica gel 60 (70–230 mesh). Thin layer chromatography (TLC) was performed on silica gel plates (Merck F245). Solvents used for extraction and chromatography such as dichloromethane, hexane, ethyl acetate and methanol were commercial grade and distilled before use while diethyl ether and chloroform were reagent grade. All reactions were carried out under positive pressure of N2 filled in rubber balloons. Milli-Q water was used in all aqueous experiments unless specified otherwise.Analytical instruments
Nuclear magnetic resonance (NMR) spectra were recorded in DMSO-d6, CDCl3, acetone-d6 and CD3CN on a Varian 400 MHz or Bruker 400 MHz or Bruker 500 MHz spectrometers. Electrospray mass spectra were acquired from a Micromass Platform Quadrupole Mass Analyser with an electrospray ion source using acetonitrile as a solvent. Absorption spectra were measured by Perkin-Elmer 35/FIAS300 and Varian Cary 50 UV-vis spectrophotometer. Fluorescence spectroscopy was performed on Agilent Cary Eclipse and Hitachi F-2500 fluorescence spectrometers.Synthesis of compound 1
A mixture of 4-ethynyl-N,N-dimethylaniline14 (146.0 mg, 1.0 mmol), 4-bromobenzaldehyde (190 mg, 1.1 mmol), PdCl2-(PPh3)2 (35.0 mg, 0.05 mmol), CuI (9.5 mg, 0.05 mmol) in THF (5 mL) was added with isopropylamine (1 mL) and the mixture was stirred at 70 °C for 15 h. The reaction mixture was then evaporated. The residue was eluted through a silica gel column by gradient from pure hexane to hexane/CH2Cl2 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (v/v) as an eluent to afford fluorophore 1 as a yellow solid (166.3 mg, 67% yield). Mp: 150–152 °C; 1H-NMR (CDCl3, 500 MHz): δ (ppm) 10.02 (s, 1H), 7.85 (d, 2H, J = 8.2 Hz), 7.63 (d, 2H, J = 8.2 Hz), 7.45 (d, 2H, J = 8.4 Hz), 6.75 (d, 2H), 3.03 (s, 6H).15
:2 (v/v) as an eluent to afford fluorophore 1 as a yellow solid (166.3 mg, 67% yield). Mp: 150–152 °C; 1H-NMR (CDCl3, 500 MHz): δ (ppm) 10.02 (s, 1H), 7.85 (d, 2H, J = 8.2 Hz), 7.63 (d, 2H, J = 8.2 Hz), 7.45 (d, 2H, J = 8.4 Hz), 6.75 (d, 2H), 3.03 (s, 6H).15
Synthesis of compound 2
Compound 1 (99.8 mg, 0.39 mmol) and indolium salt (120.5 mg, 0.40 mmol) were dissolved in 20 mL of CH2Cl2 and then AlCl3 (50 mg, 0.38 mmol) was added. The reaction mixture was stirred for 3 h at room temperature and the solvent was removed under reduced pressure. The resulting residue was purified by column chromatography on silica (from hexane to DCM/MeOH; 100:1 v/v) to give fluorophore 2 as dark violet solid (145 mg, 70%). Mp: decomposed > 250 °C; 1H-NMR (500 MHz, acetone-d6): δ (ppm) 8.40 (d, J = 20 Hz, 1H), 8.04 (d, J = 10 Hz, 2H), 7.86–7.80 (m, 1H), 7.79–7.75 (m, 1H), (m, 2H), 7.67–7.60 (m, 5H), 7.40 (d, J = 10 Hz, 2H), 6.74 (d, J = 10 Hz, 2H), 4.19 (s, 3H), 3.01 (s, 6H), 1.86 (s, 6H). 13C-NMR (500 MHz, acetone-d6): δ (ppm) 183.0, 153.7, 151.5, 144.1, 142.4, 133.3, 132.0, 130.7, 130.3, 129.7, 123.6, 123.1, 115.3(2), 112.2, 96.9, 87.8, 53.2, 39.5, 25.5, 21.7. HR-ESI-MS m/z for C29H29N2 calcd 405.2325; found 405.2322.
Photophysical property study
The stock solution of 500 μM compound 2 in 10 mM HEPES buffer pH 7.4 was prepared. The UV-vis absorption spectra of the solutions were recorded in a 1 cm quartz cell from 300 nm to 600 nm at ambient temperature. The fluorescence spectra were recorded from 380 nm to 700 nm at ambient temperature using an excitation wavelength at 370 nm. Fluorescence quantum yields of the fluorophores were determined from the slope of the plot between the integral of emission intensity against the absorbance (always kept below 0.1) in relation to quinine sulphate (ΦF = 0.54) in 0.1 M H2SO4.16 The spectra were acquired from 1–5 μM solutions, diluted from the stock solution with their respective solvents, in a 1.4 mL fluorescence quartz cell with 1 cm path length.UV-vis absorption measurements
All the UV-vis absorption spectra were acquired from aqueous solution with 10 mM HEPES buffer, pH 7.4. The UV-vis spectral changes of compound 2 (50 μM) were recorded upon the addition of sodium salts of the anion while keeping the concentration constant at 2 mM. The stock solution of α-cyclodextrin (2 mM) was prepared in HEPES buffer 7.4. The mixture between α-cyclodrextrin (400 μM), compound 2 (50 μM) and sodium salt of the anions tested (Br−, Cl−, CO32−, F−, I−, NO2−, S2−, and SO42− (1 mM) and CN− (100 μM)) were used for the UV-vis experiments.Fluorescence sensing study
The 150 mM stock solutions of the anions were prepared by dissolving their sodium salts in Milli-Q water. The anion stock solutions (10 μL) were individually mixed with the stock solution of compound 2 (500 μM, 10 μL) in HEPES buffer 7.4. The volumes of the mixtures were adjusted to 1 mL by HEPES buffer 7.4 to afford the final concentration of 50 μM for the fluorophores and 1.0 mM for the anions. In the experiments involving α-CD (400 μM), the stock solution of α-CD (2 mM) was mixed with the fluorophore stock solution (500 μM, 100 μL) prior to the addition of the anion salts. The emission spectra were recorded from 380 to 650 nm with the excitation wavelengths of 360 nm after 6 minutes of mixing under 25 °C.Paper-based sensor
The solution of compound 2 (1.0 mM) in ethanol was pipetted at 2.0 μL onto the surface of marked filter paper and allowed for air dry to generate fluorescent spots under an ordinary 20 W black light lamp. Water samples, i.e. drinking water and mineral water, spiked with sodium cyanide at various concentrations were filtered and pipetted at 2 μL on top of the spots of 2. After air drying, the images of the filter paper under the black light were photographically recorded.Results and discussion
The synthesis of the target compound 2 was done in two steps, Sonogashira coupling and Knoevenagal condensation (Scheme 1). Starting from 4-ethynyl-N,N-dimethylaniline, the Sonogashira coupling with 4-bromobenzaldehyde gave compound 1 in satisfactory yield. The Knoevenagal condensation of 1 with indolium salts afforded the target product 2 in good yield. The reaction set up was simple and the purification was easy. The structure of 2 was confirmed by 1H-NMR, 13C-NMR, and HRMS data (see ESI; Fig. S1–S3†).Compound 2 exhibited good solubility in common polar organic solvents, such as acetone, DMF, DMSO, CH3CN, THF, and EtOH but it was slightly soluble in 100% water media (Fig. S4†). The α, β, and γ-CD have been used to increase water solubility of 2.13 The ESI; Fig. S4† showed that 2 can completely solute in HEPES buffer at pH 7.4. Upon the addition of CN− (100 μM), the I/I0 of 2 (50 μM) with α-CD (400 μM) exhibited the highest ratio about 10 times as shown in ESI; Fig. S5.† A solution of 2 mixed with α-CD (400 μM) in HEPES buffer at pH 7.4 was light reddish orange with two distinct absorption bands around 360 and 450 nm (Fig. 1). The emission maxima appeared around 450 and 495 nm with the fluorescence quantum efficiency of 0.025 (quinine sulfate = 0.54 was used as reference).16 The low quantum efficiency is probably due to the intramolecular charge transfer (ICT) from the strong electron donor amino group to the strong electron acceptor indolium group via its π-conjugated system resulting in excited ICT state with much smaller band gap.17 Additionally, intermolecular H-type π–π interaction may also contribute to the self-quenching effect.18
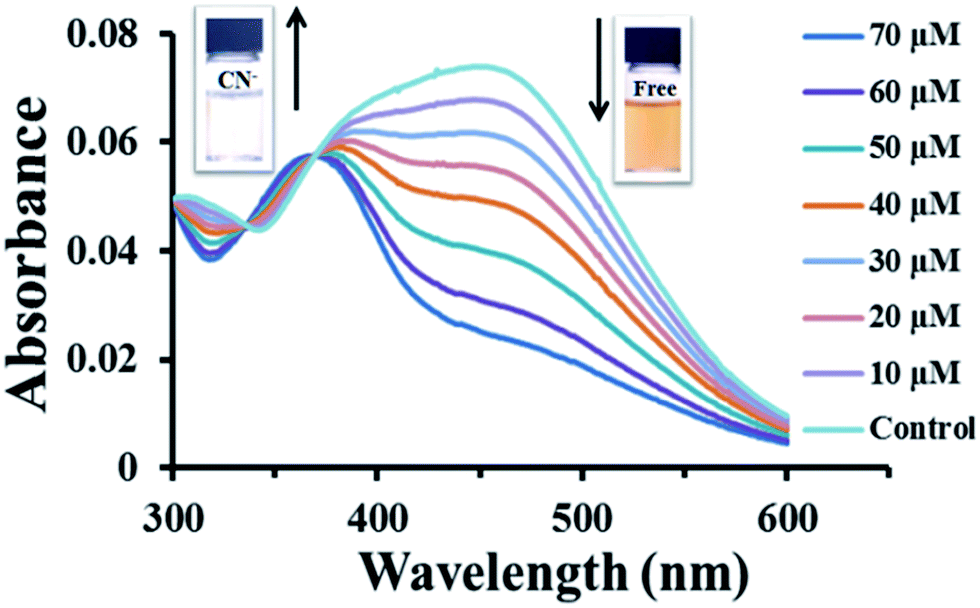 | ||
| Fig. 1 UV-vis spectra of 2 (50 μM) and cyclodextrin (400 μM) in 10 mM HEPES buffer pH 7.4 in the presence of different concentrations of CN−. | ||
Upon the addition of sodium cyanide to the solution of 2 in 10 mM HEPES buffer pH 7.4, the absorption band at 450 nm decreased with an increase of a new band at 360 nm (Fig. 1). The color of the solution also changed from orange to colorless which was easily observed by naked eye. A large hypsochromic shift of 90 nm in the electronic absorption peak is associated with the nucleophilic attack of CN− to the iminium carbon of the indolium group.11a,b This addition reaction disrupts the electron accepting ability of the indolium group as well as electron delocalization within its π-conjugated system. Job's plot analysis of the fluorescence responses revealed that the 2 formed 1:1 complexes with CN−. The analysis mechanism of the method was formed stoichiometrically (Fig. 2).
 | ||
| Fig. 2 Job's plot of fluorescence responses of 2 (50 μM) mixed cyclodextrins (400 μM). To various mole faction of CN− in HEPES buffer pH 7.4. | ||
Compound 2 showed weak emission (ΦF = 0.025) with a maximum at 595 nm. Upon the addition of sodium cyanide, the fluorescence of 2 dramatically and rapidly increased with the blue-green emission and maximum emission wavelength at 450 nm (Fig. 3) at 6 minutes under 25 °C (ESI; Fig. S6†). The quantum efficiency of the cyanide adduct improved to 0.34 (ESI; Table S1†). The fluorescence turn-on response ratio (I/I0) linearly increased with the cyanide concentration in the range of 0–80 μM (ESI; Fig S7†). The detection limit of cyanide determined at S/N = 3 was 1.3 μM below the World Health Organization (WHO) limit of 1.9 μM allowed in drinkable water.2,19
 | ||
| Fig. 3 Emission spectra of 2 (50 μM) and cyclodextrin (400 μM) in response to addition of sodium cyanide at various concentrations in 10 mM HEPES buffer pH 7.4 (λex = 360 nm). | ||
1H-NMR titration was used to verify that nucleophilic addition of CN− to the imminium carbon of the indolium group (Fig. 4) really participates in the cyanide sensing mechanism of 2. The spectra in Fig. 5 show that the signal at 6.42 ppm corresponding to the g′ proton grows in expense of the signal of g vinylic proton at 8.40 ppm upon the addition of sodium cyanide. Meanwhile, the signals of the aromatic protons also displayed large magnetically shielded shifts compared with those of 2 in good agreement with the loss of conjugated electron withdrawing character of the indolium group.11 Additionally, the formation of cyanide with 2 was confirmed by high resolution mass HRMS where a peak at m/z 432.2400 corresponding to the cyanation adduct (Fig. S8†).
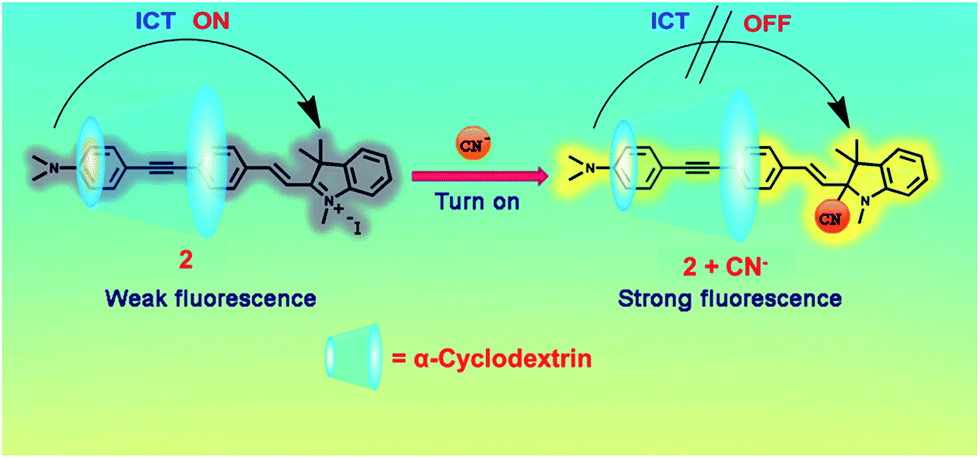 | ||
| Fig. 4 Proposed structure of inclusion complexes between 2 with α-CD and cyanide sensing mechanism. | ||
 | ||
| Fig. 5 1H-NMR spectra (500 MHz, CD3OD) of 2 only (0.9 mM) in the absence and presence of cyanide. | ||
To evaluate the selectivity of 2 toward various anions i.e. Br−, Cl−, CO32−, F−, I−, NO2−, S2− and SO42− were examined in parallel to CN− using their sodium salts. As shown in Fig. 6, only sodium cyanide induced the color disappearance and blue emission of the original orange color of 2. Therefore, 2 has much higher selectivity for CN− than other anions. Notably, S2− also induced some color fading, probably the similar addition mechanism of S2− to 2,11c but without apparent fluorescence emission. The results suggested that a selective naked eye detection of cyanide with 2 was possible.
 | ||
| Fig. 6 Photographs of 2 (100 μM) and cyclodextrin (800 μM) in HEPES buffer (10 mM) pH 7.4 in the presence of sodium salt of anion (1.0 mM) taken under (a) room light and (b) black light. | ||
To better evaluate the interference test of 2 mixed α-CD (400 μM), ratiometric absorption (A360/A450) and fluorescence (I/I0) changes caused by the addition of other anions. Including with the interfering anion ions at 10 times concentration did not show any significant ratiometric absorption of all anion ions tested on 2 (Fig. 7a). The fluorescence enhancement ratio (I/I0) also showed high selectivity toward CN− and low interference from other types of anions including S2− (Fig. 7b).
 | ||
| Fig. 7 Bar charts of (a) ratiometric absorption (A360/A450) and (b) fluorescence intensity ratio (I/I0) of 2/α-CD (50/400 μM). Orange bars represent the addition of a single analyte, red bars exhibit the present of CN− (100 μM) in the mixture of 10 mM HEPES buffer (pH 7.4) and other interfering anions (1 mM). | ||
Compound 2 was prepared as a solid state sensor by drop casting on a strip of filter paper from its ethanol solution (100 μM, 2 μL). The test strip was dropped with sodium cyanide solution (2 μL) with various concentrations. Visualization of the dry test strip under black light (20 W) showed intense blue fluorescence when the concentration of sodium cyanide solution was 40 μM or higher (Fig. 8). The results can be translated to the detection ability of 5 ng of cyanide per square millimeters of the testing area.
 | ||
| Fig. 8 Photographic images of 2 paper-based detection of CN− under a 20 W black light (scale bar = 7 mm). | ||
The present method was applied to determine cyanide ion in the real water samples i.e. drinking water, mineral water and brine (DI water with 10% NaCl). The water samples were buffered with 10 mM HEPES buffer, pH 7.4 and spiked with known amount of sodium cyanide (10 and 20 μM). The analysis of each sample was carried out in three replicates. Table 1 demonstrates that cyanide ion was determined in with a satisfactory analytical accuracy (<10% error) and precision (RSD ≤ 5%) although the sample high concentration of brine gave a little higher positive error values. The results confirmed the accuracy and reproducibility of this sensing system. The method is also relatively simple and economical as it has high tolerance to other inferring ions.
| Samples | Spiked (μM) | Found (μM) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| a Mean ± SD. | ||||
| Drinking water | 10.0 | 9.78 ± 0.21 | 97.84 ± 2.85 | 2.16 |
| 20.0 | 20.45 ± 0.27 | 102.72 ± 2.50 | 1.78 | |
| Minerals water | 10.0 | 9.92 ± 0.13 | 99.25 ± 0.89 | 0.85 |
| 20.0 | 19.56 ± 0.41 | 97.83 ± 4.92 | 2.42 | |
| 10% NaCl (aq.) | 10.0 | 10.97 ± 0.52 | 109.71 ± 5.45 | 3.64 |
| 20.0 | 21.15 ± 0.29 | 105.75 ± 3.12 | 2.18 | |
In comparison with other sensing materials used for CN− detection in aqueous solution, the CN− detection limit of 2 is one of the lowest as shown in Table 2. To the best of our knowledge, this work is also one of the most sensitive and convenient techniques for the naked eye detection of CN− (Table 2).
| Sensing materials | Signal detected | Solvent | Detection limit | Ref. |
|---|---|---|---|---|
| Boradiazaindacene | Turn-on emission | CH3CN | 30 μM | 20 |
| Coumarin salicylaldehyde | Turn-on emission | 40% DMSO in HEPES buffer pH 7.4 | 10 μM | 8a |
| Dicyano-vinyl polymer | Turn-on emission | DMF | 14 μM | 21 |
| Benzofuranzan | Turn-on emission | 95% CH3CN/5% H2O | 1.5 μM | 22 |
| Azobenzylmalono-nitrile | Absorption | CH3CN | 1.1 μM | 23 |
| Quinolinium bromide | Absorption | EtOH | 1.9 μM | 24 |
| Compound 2 | Turn-on emission (visual observation) | 100% HEPES buffer, pH 7.4 paper | 1.3 μM | This work |
| 4.59 ng mm−1 |
Conclusions
In summary, the study on fluorophores 2 containing phenylene-ethynylene fluorescent units with indolium terminal as the most sensitive and selective sensor for CN− detection is highly reactive to CN−. The detection limit was estimated to be 1.3 μM in aqueous media. The ratio colorimetric and fluorometric sensing behaviour of 2 for CN− was from the reduced π-conjugation and block ICT process induced by CN− upon the addition of 2. Effectively, the probe 2 could be applied to detect CN− in drinking water with satisfactory results. The test strip containing 2 also exhibits a good sensitivity to CN− ion at 4.59 ng mm−1 level. The probe 2 was constructed by taking advantages of the cyanide sensor. In addition, the probe could serve as practical colorimetric sensors for “in-the-field” measurement, which did not require any additional equipment.Acknowledgements
The authors gratefully thank the Thailand Research Fund (MRG5680031) and KMUTT Research Fund. M. S. would also like to acknowledge the financial supports from the Thailand Nanotechnology Center (NANOTEC, NSTDA), the IIAC Chulalongkorn University Centenary Academic Development Project, the National Research University Project of Thailand, Office of the Higher Education Commission (AM1006A), the Project for Establishment of Comprehensive Centre for Innovative Food, Health Products and Agriculture supported by the Thai government stimulus package 2 (TKK2555, SP2), the Ratchadaphiseksomphot Endowment Fund (90th Anniversary CU Fund).Notes and references
- (a) K. W. Kulig, Cyanide Toxicity, U.S. Department of Health and Human Services, Atlanta, GA, 1991 Search PubMed; (b) F. J. Baud, Hum. Exp. Toxicol., 2007, 26, 191 CrossRef CAS PubMed; (c) R. Yang, W. Wu, W. Wang, Z. Li and J. Qin, Chem. Phys., 2010, 211, 18 CAS; (d) X. Chen, S. W. Nam, G. H. Kim, N. Song, Y. Jeong, I. Shin, S. K. Kim, J. Kim, S. Park and J. Yoon, Chem. Commun., 2010, 8953 RSC.
- Guidelines for Drinking-Water Quality, ed. J. K. Fawell, J. R. Hickman, U. Lurid, B. Mintz, E. B. Pike, H. Galal-Gorchev, R. Helmer, X. Bonnefoy and O. Espinoza, World Health Organization, Geneva, 1996 Search PubMed.
- (a) G. Qian, X. Z. Li and Z. Y. Wang, J. Mater. Chem., 2009, 19, 522 RSC; (b) L. H. Peng, M. Wang, G. H. Zhang, D. Q. Zhang and D. B. Zhu, Org. Lett., 2009, 11, 1943 CrossRef CAS PubMed.
- (a) J. E. Poulton, Plant Physiol., 1990, 94, 401 CrossRef CAS PubMed; (b) J. Vetter, Toxicon, 2000, 38, 11 CrossRef CAS.
- (a) R. D. Rocklin and E. L. Johnson, Anal. Chem., 1983, 55, 4 CrossRef CAS; (b) P. R. Haddad and N. E. Rochester, J. Chromatogr. A, 1988, 439, 23 CrossRef CAS; (c) M. Dyrby, S. B. Engelsen, L. Norgaard, M. Bruhn and L. Lundsberg-Nielsen, Appl. Spectrosc., 2002, 56, 579 CrossRef CAS; (d) J. Jo and D. Lee, J. Am. Chem. Soc., 2009, 131, 16283 CrossRef CAS PubMed; (e) N. Kumari, S. Jha and S. Bhattacharya, Chem.–Asian J., 2014, 9, 830 CrossRef CAS PubMed.
- (a) Z. C. Xu, X. Q. Chen, H. N. Kim and J. Y. Yoon, Chem. Soc. Rev., 2010, 39, 127 RSC; (b) F. Wang, L. Wang, X. Chen and J. Yoon, Chem. Soc. Rev., 2014, 43, 4312 RSC.
- (a) R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635 CrossRef CAS PubMed; (b) M. Jamkratoke, V. Ruangpornvisuti, G. Tumcharern, T. Tuntulani and B. Tomapatanaget, J. Org. Chem., 2009, 74, 3919 CrossRef CAS PubMed.
- (a) K. S. Lee, S. J. Kim, J. H. Kim, I. Shinand and J. I. Hong, Org. Lett., 2008, 10, 49 CrossRef CAS PubMed; (b) N. Niamnont, A. Khumsri, A. Promchat, G. Tumcharern and M. Sukwattanasinitt, J. Hazard. Mater., 2014, 280, 458 CrossRef CAS PubMed.
- (a) X. Cheng, R. Tang, H. Jia, J. Feng, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4387 CrossRef CAS PubMed; (b) P. B. Patia and S. S. Zade, RSC Adv., 2013, 3, 13457 RSC.
- (a) M. Tomasulo and F. M. Raymo, Org. Lett., 2005, 7, 4633 CrossRef CAS PubMed; (b) V. Kumar, H. Rana and M. P. Kaushik, Analyst, 2011, 136, 1873 RSC; (c) L. Yang, X. Li, J. B. Yang, Y. Qu and J. L. Hua, ACS Appl. Mater. Interfaces, 2013, 5, 1317 CrossRef CAS PubMed; (d) S. S. Razi, R. Ali, P. Srivastava, M. Shahida and A. Misra, RSC Adv., 2014, 4, 22308 RSC; (e) R. K. Konidena and K. R. J. Thomas, RSC Adv., 2014, 4, 22902 RSC; (f) M. J. Peng, Y. Guo, X. F. Yang, F. Suzenet, J. Li, C. W. Li and Y. W. Duan, RSC Adv., 2014, 4, 19077 RSC.
- (a) H. J. Kim, K. C. Ko, J. H. Lee, J. Y. Lee and J. S. Kim, Chem. Commun., 2011, 47, 2886 RSC; (b) X. Lv, J. Liu, Y. Liu, Y. Zhao, Y. Q. Sun, P. Wang and W. Guo, Chem. Commun., 2011, 47, 12843 RSC; (c) Y. Zhang, D. Yu and G. Feng, RSC Adv., 2014, 4, 14752 RSC; (d) S. Wang, H. Xu, Q. Yang, Y. Song and Y. Li, RSC Adv., 2015, 5, 47990 RSC.
- (a) S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed; (b) N. Niamnont, R. Mungkarndee, I. Techakriengkrai, P. Rashatasakhon and M. Sukwattanasinitt, Biosens. Bioelectron., 2010, 26, 863 CrossRef CAS PubMed.
- W. Siripornnoppakhun, N. Niamnont, A. Krumsri, G. Tumcharern, T. Vilaivan, P. Rashatasakhon and M. Sukwattanasinitt, J. Phys. Chem. B, 2012, 116, 12268 CrossRef CAS PubMed.
- N. Niamnont, W. Siripornnoppakhun, P. Rashatasakhon and M. Sukwattanasinitt, Org. Lett., 2009, 11, 2768 CrossRef CAS PubMed.
- F. Zhou, S. Jingyin, Y. Yubin, Z. Jianzhang, G. Huimin, L. Xiaolian, J. Shaomin and Z. Zongying, Eur. J. Org. Chem., 2011, 4773 CAS.
- S. Fery-Forgues and D. Lavabe, J. Chem. Educ., 1999, 76, 1260 CrossRef CAS.
- S. Masuo, H. Yoshikawa, T. Asahi and H. Masuhara, J. Phys. Chem. B, 2003, 107, 2471 CrossRef CAS.
- (a) Q. Chu and Y. Pang, Macromolecules, 2005, 38, 517 CrossRef CAS; (b) J. A. Richard, M. Massonneau and P. Y. Renard, Org. Lett., 2008, 10, 4175 CrossRef CAS PubMed.
- M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414 CrossRef CAS.
- Z. Ekmekci, M. D. Yilmaz and U. D. Akkaya, Org. Lett., 2008, 10, 461 CrossRef CAS PubMed.
- X. Wu, B. Xu, H. Tong and L. Wang, Macromolecules, 2011, 44, 4241 CrossRef CAS.
- Z. Liu, X. Wang, Z. Yang and W. He, J. Org. Chem., 2011, 76, 10286 CrossRef CAS PubMed.
- X. Cheng, Y. Zhou, J. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 2133 CAS.
- H. M. Nie, C. B. Gong, Q. Tang, X. B. Maa and C. F. Chow, Dyes Pigm., 2014, 106, 74 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectroscopic data. See DOI: 10.1039/c5ra10273a |
| This journal is © The Royal Society of Chemistry 2015 |