
Fast diffusion supercapacitors via an ultra-high pore volume of crumpled 3D structure reduced graphene oxide activation†
Abstract
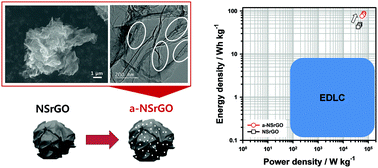
In order to obtain a high performance supercapacitor, there are several factors that must be achieved including a high specific surface area (SSA), high electrical conductivity, and a high diffusion rate of the electrolyte due to an appropriate pore volume. Herein, we report a high performance supercapacitor using activated non-stacked reduced graphene oxide (a-NSrGO) that has a high SSA (up to 999.75 m2 g−1) with intrinsic high graphene conductivity (1202 S m−1) and fast diffusion of the electrolyte. Due to a high total pore volume (5.03 cm3 g−1) and a wide pore size distribution from macro- to micropores (main pore width: 0.61 – 0.71 nm) in the a-NSrGO sheets, the as-prepared a-NSrGO electrode shows high specific capacitance (105.26 F g−1) and a short relaxation time (τ0 = 1.5 s) in a propylene carbonate (PC)-based organic electrolyte. A maximum energy density of 91.13 W h kg−1 and a power density of 66 684.73 W kg−1 were estimated in a fully packaged coin cell. The high performance of the a-NSrGO supercapacitors is attributed to their specific appearance and enlarged pore distribution with high SSA.
 Please wait while we load your content...
Please wait while we load your content...

