
An extremely highly recoverable clay-supported Pd nanoparticle catalyst for solvent-free Heck–Mizoroki reactions†
Alejandro V. Martínez,
Alejandro Leal-Duaso,
José I. García and
José A. Mayoral*
Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) and Department of Organic Chemistry, Facultad de Ciencias. CSIC-Universidad de Zaragoza, C/Pedro Cerbuna, 12, E-50009 Zaragoza, Spain. E-mail: mayoral@unizar.es
First published on 6th July 2015
Abstract
Palladium nanoparticles supported on LAPONITE®, a synthetic clay, are shown to constitute a highly robust and stable catalytic system for Heck–Mizoroki reactions carried out under solventless conditions. Both the preparation and the use of the catalyst are straightforward, allowing easy separation of the reaction products in pure form. In the case of the reaction between butyl acrylate and iodobenzene, up to 50 catalyst cycles can be performed before observing a noticeable decrease in activity. After reactivation of the catalyst by calcination, it can be further used for at least an additional 25 reactions. The stability of the catalyst seems to be related to the electronic density of the reacting alkene, as illustrated by the results obtained using the same catalyst with other alkenes.
Introduction
Palladium catalysts play a decisive role in modern organic synthesis due to their ability to promote a variety of bond-forming reactions. Palladium-catalyzed Suzuki–Miyaura,1 Heck–Mizoroki2 and Sonogashira–Hagihara3 coupling reactions are powerful tools for the selective formation of C–C bonds in specialty industries.4 In recent surveys, it has been described that these kinds of palladium catalyzed reactions have gained importance within the category of C–C bond forming reactions used in the synthesis of drug candidate molecules, passing from 22% in 2006 to 63% in 2011.5,6 Because of the limited availability and high price of palladium, it is important to develop palladium catalysts with excellent recyclability and low metal leaching.7 One of the strategies to achieve this goal is the immobilization of Pd(II) complexes onto insoluble supports. In this regard different complexes have been grafted onto inorganic supports, organic polymers and membranes.8–11 Recently, a molecular Pd(II) complex incorporated into a MOF has been shown to be a highly active single-site heterogeneous catalyst for the activation of C–Cl bonds.12 A palladium complex immobilized on the surface of gold nanoparticles is one of the catalysts with the highest recoverability described to date for these reactions:13 with 8% mol of the Pd(II) complex the catalyst was used fifteen times in the reaction between iodobenzene and n-butyl acrylate with yields decreasing only from 99% to 79%.A second strategy is the use of a solvent allowing the easy separation of the phase containing the palladium complex. In a recent example a water-soluble Pd–salen complex has been used in the reactions between iodobenzene and several alkenes in water.14 In the reaction with styrene, the catalyst was recovered five times with yields decreasing from 98% to 92%.
An alternative to the use of grafted molecular complexes is the use of Pd metal catalysts15 and, in particular, Pd nanoparticles (PNP). Some of the works in this field have described the use of supported PNP as recoverable catalysts for Suzuki–Miyaura reactions. Thus, PNP supported on microporous polymers have been used up to nine times in the reaction between bromobenzene and phenylboronic acid.16 Using Fe3O4–SiO2 nanoparticles functionalized with chlorodiphenylphosphine as the support, the resulting supported nanopalladium catalyst was reused six times keeping a high activity and with only 2.5% leaching of palladium.17 PNP have also been supported on partially reduced graphene oxide nanosheets18 and on single- and multi-walled carbon nanotubes.19 The latest support led to the best results in terms of catalyst reuse, the yield decreasing from 100% to 70% after ten reactions and to 52% in the eleventh use. Given the small amount of Pd leached, agglomeration of nanoparticles is suggested as the responsible for catalyst deactivation. Similar behavior, with a decrease in yield from 100% to 62% after nine cycles, was observed using graphene as the catalyst support.20 Very recently carbon-supported PNP have been used ten times in Suzuki–Miyaura reactions carried out in aqueous media.21 The highest catalyst stability reported to date has been reached with PNP embedded in polymer thin films. With this catalyst a high activity is retained beyond thirty cycles in the prototypical reaction between iodobenzene and phenylboronic acid in ethanol, using K2CO3 as the base.22
Regarding the Heck–Mizoroki reaction, PNP immobilized on magnetic nanoparticles of Fe3O4 functionalized with agarose were used six times in the reaction of iodobenzene with n-butyl acrylate with yields decreasing from 94% to 86%.23 Using magnetic Fe3O4–SiO2 nanoparticles functionalized with chlorodiphenylphosphine as the support, the catalyst was used six times keeping a high activity and with only 4.5% total leaching of palladium.17 When the PNP were deposited on polylactic-grafted carbon nanotubes the yield decreased from 61.5% to 59.5% in the reaction of bromobenzene with methyl acrylate.24 The pectin-supported catalyst has been studied in the reaction of iodobenzene with n-butyl acrylate at 140 °C. After six reactions the time needed to reach 93% yield increased from 5 to 20 minutes with 4% total Pd leaching.25
Regarding the use of purely inorganic supports it is worth nothing the seminal work from Choudary et al. who described the use of layered double hydroxides-supported PNP as catalysts for several cross-coupling reactions, although recycling experiments were very limited.26 In another pioneer work27 diatomite was used as the support of the PNP, and both Suzuki–Miyaura and Heck–Mizoroki reactions were studied. The catalyst was used six times, but an important increase of the reaction time was necessary in the last two cycles to keep high yields. Given the noticeable metal leaching, the diatomite-supported PNP were considered as a palladium source. Recently we have described the use of PNP immobilized on ionic liquids films supported on LAPONITE® clay as highly reusable catalysts for the Heck–Mizoroki reaction.28 Closely related catalytic systems have also been tested in connection with the microwave activation of this reaction.29 The results obtained pointed out to investigate whether the ionic liquid is a key element for PNP support and what is the influence of the clay on the recoverability of the catalysts in solvent-free Heck–Mizoroki reactions activated under conventional heating.
Results and discussion
Catalyst synthesis and characterization
LAPONITE®-supported Pd nanoparticles (PNP) were prepared using a straightforward procedure, according to previously described methods.28,30,31Polyvinylpyrrolidone (PVP)-stabilized nanoparticles were obtained by reduction of H2PdCl4 with ethanol, followed by impregnation of the LAPONITE® clay with a dichloromethane suspension of the PNP, and subsequent solvent evaporation to lead to the supported PNP as a free-flowing powdery grey solid. The surface area of the resulting solid, determined by N2 adsorption by applying the BET theory (301.9 m2 g−1, pore volume 0.264 cm3 g−1), is not very different from that of the pure LAPONITE® support (348.2 m2 g−1, pore volume 0.268 cm3 g−1). The palladium content was determined in several samples by ICP as being in the range 2.65 to 3.05 × 10−3 mmol g−1, not far from the theoretical value arisen from the preparation procedure (3 × 10−3 mmol g−1). PNP size distribution was determined by the analysis of the TEM images (Fig. 1). The size range is 0.5–3.5 nm, with a typical value of 1.8 ± 0.6 nm in the case of the ethanolic colloidal suspension of PVP-stabilized PNP, whereas the typical size of the LAPONITE®-supported PNP is bigger, with a size range of 1–14 nm and a typical value of 6.3 ± 1.8 nm. This indicates a growing of the PNP during the process of deposition on the support.
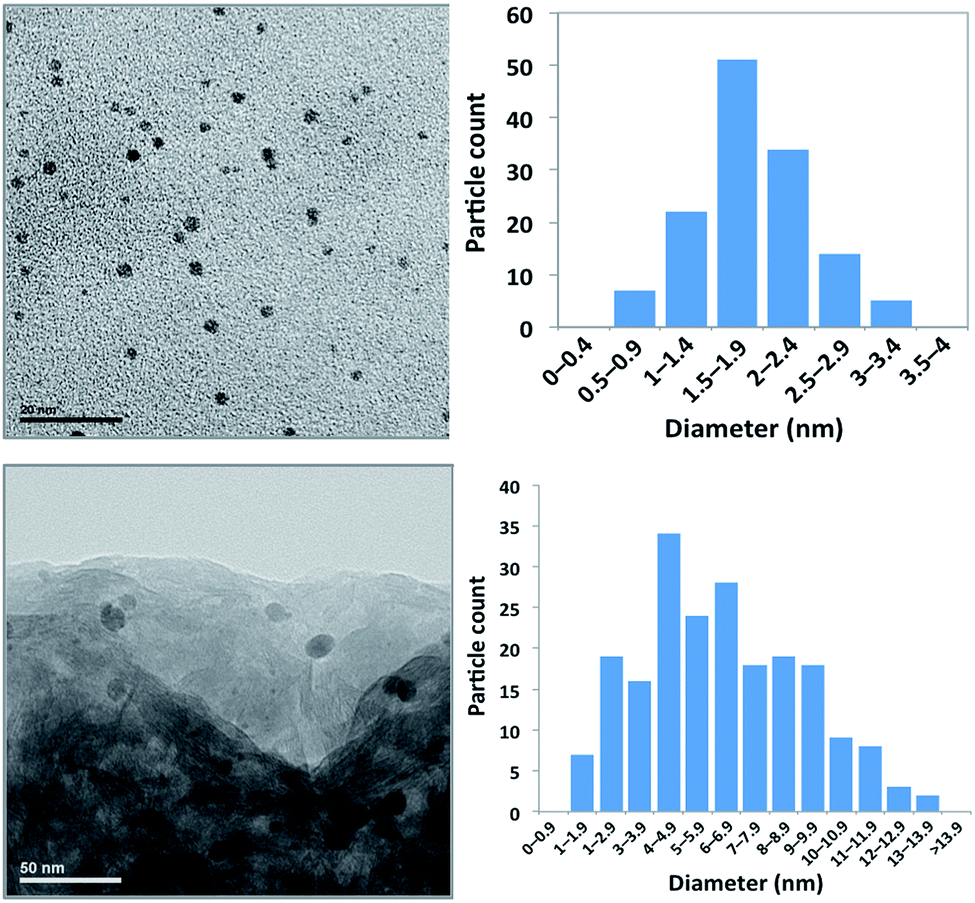 | ||
| Fig. 1 TEM micrographs and size distribution of the Pd nanoparticles (PNP) used in this study. Upper view: colloidal suspension of PVP-stabilized PNP. Lower view: PVP-stabilized PNP supported on LAPONITE®. | ||
Catalytic tests in the benchmark reaction
The supported catalyst was tested in the benchmark reaction of iodobenzene with n-butyl acrylate, in the presence of triethylamine and without using any additional solvent (Scheme 1). Under these conditions, reagents are completely embedded into the solid, which keeps a free-flowing powdery aspect. The amount of solid catalyst used was chosen to formally have 1% mol Pd in the reaction vessel, although of course, much less Pd is actually available as catalyst, since most of the Pd atoms are buried inside the nanoparticles. At the end of the reaction the solid was thoroughly extracted with hexane and the extract analyzed by gas chromatography. The solid was dried and reused under the same conditions in subsequent reaction cycles, showing an excellent recyclability. Table 1 gathers the conversions and yields obtained in the first 50 uses of this catalytic system.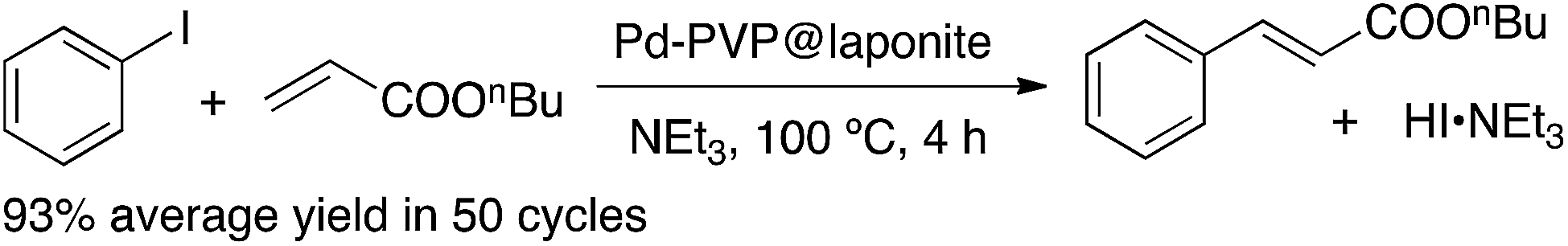 | ||
| Scheme 1 Benchmark Heck–Mizoroki reaction between iodobenzene and butyl acrylate, catalyzed by LAPONITE®-supported palladium nanoparticles in solventless conditions. | ||
| Run | Conversionc,d (%) | Yieldc,d (%) | Isolated yield (%) |
|---|---|---|---|
| a Using a 1% Pd total molar ratio, at 100 °C during 4 hours.b Complete results available in the ESI.c Determined by GC using n-decane as the internal standard. Regio- and stereoselectivity to butyl (E)-cinnamate was 100% in all cases.d Results of the first 30 runs are the average of two parallel reactions. | |||
| 1d | 95 | 94 | 90 |
| 2 | 94 | 94 | 90 |
| 3 | 100 | 100 | 99 |
| 4 | 100 | 100 | 94 |
| 5 | 100 | 100 | 94 |
| 6 | 100 | 100 | 95 |
| 7 | 100 | 100 | 95 |
| 8 | 100 | 100 | 92 |
| 9 | 100 | 100 | 88 |
| 10 | 97 | 97 | 93 |
| 15 | 100 | 100 | 97 |
| 20 | 94 | 94 | 95 |
| 25 | 94 | 94 | 96 |
| 30 | 96 | 96 | 83 |
| 35 | 78 | 76 | 67 |
| 40 | 94 | 94 | 92 |
| 45 | 64 | 62 | 57 |
| 50 | 86 | 85 | 80 |
As can be seen, initial reactions lead to high conversions and yields and the catalyst show a high activity during 50 runs. The average conversion and yield percentages are 93 ± 1.8 and 93 ± 1.7, respectively. Isolated yields, on the other hand, display a somewhat higher variability (average value 89 ± 3.3%), especially when the catalyst has already been used more than 30 times, which may be due to the changes in the nature of the remaining solid. In fact, the overall mass of the solid increases upon recovery since the concomitant byproduct (triethylammonium iodide) does not dissolve in hexane and hence remains deposited on the solid. This is illustrated by the thermogravimetric analysis of the catalyst used 50 times, which shows that there is 85% weight loss at 274 °C (Fig. 2).
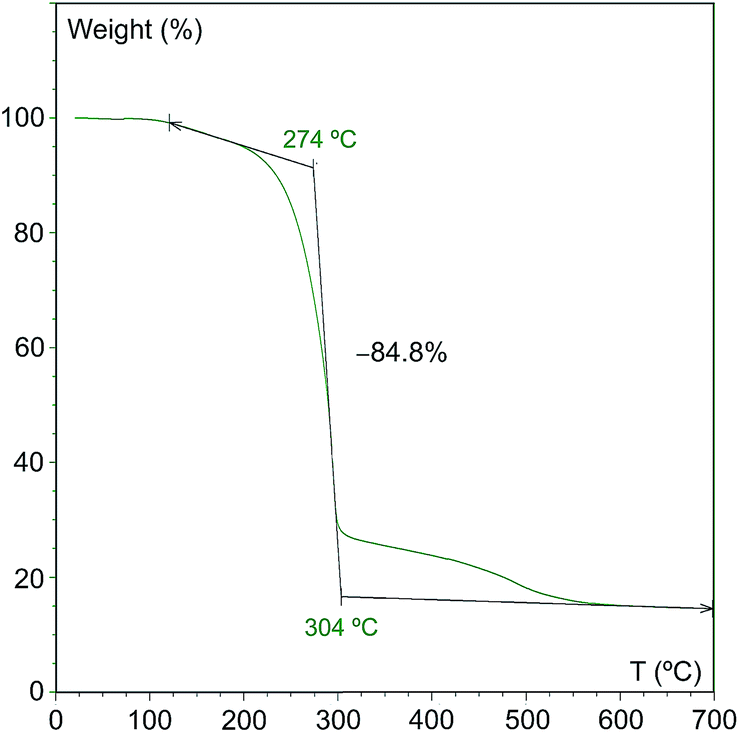 | ||
| Fig. 2 Thermogravimetric analysis of a sample of the catalyst used in 50 consecutive runs. | ||
Furthermore, elemental analysis shows a considerable increase in C and N with regard to the freshly prepared catalyst, reaching a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar proportion. All these results confirm the deposition of triethylammonium iodide on the clay. These changes in the nature of the solid may interfere with the extraction process, thus justifying the variability observed in the last reuses, where a low yield in one cycle is followed by a high yield in the next cycle, most probably due to an incomplete product extraction.
:1 molar proportion. All these results confirm the deposition of triethylammonium iodide on the clay. These changes in the nature of the solid may interfere with the extraction process, thus justifying the variability observed in the last reuses, where a low yield in one cycle is followed by a high yield in the next cycle, most probably due to an incomplete product extraction.
We also examined Pd leaching by comparing initial and final palladium contents in samples previously calcined to remove organic compounds. To avoid any interference of the calcination process we have also determined the Pd/Mg ratio (LAPONITE® is a synthetic sodium lithium magnesium silicate with a definite composition) as a probably more reliable determination of Pd leaching. As can be seen in Table 2, entries 1–4, the most important Pd loss takes place in the first catalyst use (ca. 9% Pd leaching), whereas in the two subsequent reactions the loss is much more modes (ca. 1% each). In the catalyst samples used in 50 runs (Table 2, entry 5) the Pd leaching is ca. 19%, indicating that Pd leaching is greatly reduced as the recycled catalyst use increases. The comparison of these results with those obtained using ionic liquid films on LAPONITE® as the support28 shows that the present catalyst preparation and operation is really useful to reduce Pd leaching, which is probably also one of the reasons for the high catalyst stability and durability observed.
| Entry | Run | Pd (mg g−1) | Pd/Mg ratio (×103) | Pd leachingb (%) | Pd leachingc (%) |
|---|---|---|---|---|---|
| a All the catalyst samples were calcined at 550 °C under air prior analyses. Pd analyses carried out with ICP-MS and Mg analyses with ICP-OES.b Calculated from the absolute Pd content.c Calculated from the Pd/Mg ratio. | |||||
| 1 | 0 | 0.3050 | 1.848 | — | — |
| 2 | 1 | 0.2778 | 1.709 | 8.9 | 7.6 |
| 3 | 2 | 0.2765 | 1.691 | 9.4 | 8.5 |
| 4 | 3 | 0.2724 | 1.670 | 10.7 | 9.7 |
| 5 | 50 | 0.2458 | 1.509 | 19.4 | 18.4 |
| 6 | 75 | 0.2372 | 1.442 | 22.2 | 22.0 |
Both leaching and sintering of nanoparticles have been claimed as the main reasons for deactivation of Pd-metal catalysts used to promote Heck–Mizoroki reactions.15 To investigate the changes experienced by the catalyst after 50 uses we have examined catalyst samples by electron microscopy (FESEM and TEM). The comparison of the SEM micrographs of samples of freshly prepared and used catalyst (Fig. 3) reveals the significant decrease of the LAPONITE® particle size, probably due to extensive delamination of its structure, as revealed in the enlarged images.
 | ||
| Fig. 3 FESEM micrographs of the freshly prepared catalyst (up) and the catalyst after 50 runs (down). | ||
The analysis of the TEM micrographs reveals that the size of the PNP in the recovered catalyst is smaller than that measured in the freshly prepared one, with a typical value of 2.9 ± 1.2 nm (median value of 2.8 nm) (Fig. 4). This represents a clear decrease of the PNP size along the reuse of the supported catalyst, which points to a role of nanoparticles as palladium reservoirs. In this context, there is still an open discussion about the nature of the true catalytic species involved in the catalysis of cross-coupling reactions by PNP. There is some evidence supporting the formation either of coordinatively unsaturated Pd atoms or anionic halide complexes, which would be the true catalytic species, and that would redeposit onto the support at the end of the reaction.32 In our case, since the nanoparticles are deposited onto a solid support and the reaction is carried out in solventless conditions, it is difficult to ascertain the true nature of the catalytic species, as well as the form in which palladium atoms are leached from the catalyst. To investigate the heterogeneous character of the reaction, a freshly prepared catalyst was used, after 1.5 hours the reaction was cooled and the solid extracted with hexane. The solvent was eliminated under reduced pressure and, after the addition of a new amount of triethylamine, the reaction was continued for six days. In these conditions, yield increased from 16% in the first extract to 18% in the extract taken the sixth day. The same experiment was repeated after three hours of reaction with yields going from 87% to 88% after the same period. These filtration experiments point to the catalytic inactivity of the possible palladium species leached.
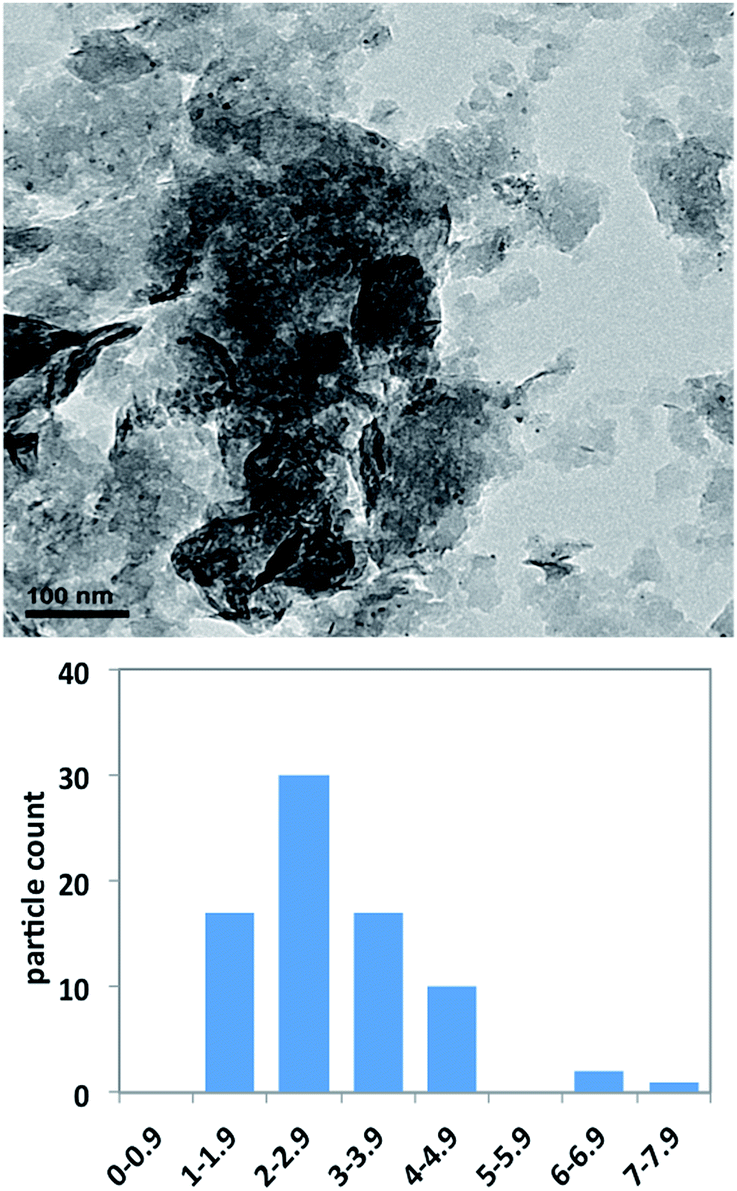 | ||
| Fig. 4 FESEM micrographs of the freshly prepared catalyst (up) and the catalyst after 50 runs (down). | ||
It is possible to suggest a reason for the high stability observed, unprecedented for a PNP-supported catalyst, namely, the use of a cationic support and the absence of a solvent favor the deposition of the concomitant ionic product and this ammonium salt protects the catalyst from both sintering and leaching. An additional advantage of this situation is that the whole byproduct remains adsorbed on the clay, and therefore additional product purification steps are avoided. After fifty cycles 85% of the solid weight corresponds to the ammonium salt, as already mentioned, which renders the catalyst difficult to handle. For this reason we decided to remove all this organic matter by calcination of the recovered solid under air for 10 hours at 550 °C, the temperature at which no additional weight loss is observed in the TGA experiments (Fig. 2). Next, we reused the resulting solid as catalyst under the same conditions (Table 3), which constitutes an additional test of catalyst stability, since the calcination under oxidant conditions could result in its total deactivation.
| Catalyst | Run | Conversionb (%) | Yieldb (%) | Isolated yield (%) |
|---|---|---|---|---|
| a Reactions carried out at 100 °C for 4 hours.b Determined by GC using n-decane as the internal standard. Regio- and stereoselectivity to butyl (E)-cinnamate was 100% in all cases.c Calcined at 550 °C under air stream for 10 hours. | ||||
| 1st calcinationc | 51 | 100 | 100 | 91 |
| 52 | 98 | 96 | 94 | |
| 53 | 96 | 96 | 95 | |
| 54 | 100 | 100 | 100 | |
| 55 | 90 | 89 | 76 | |
| 56 | 97 | 97 | 95 | |
| 57 | 100 | 100 | 97 | |
| 58 | 15 | 14 | 14 | |
| 2nd calcinationc | 59 | 100 | 100 | 99 |
| 60 | 100 | 100 | 93 | |
| 61 | 99 | 99 | 96 | |
| 62 | 99 | 99 | 99 | |
| 63 | 100 | 100 | 81 | |
| 64 | 100 | 100 | 74 | |
| 65 | 5 | 1 | 1 | |
| 3rd calcinationc | 66 | 100 | 100 | 81 |
| 67 | 89 | 88 | 82 | |
| 68 | 100 | 100 | 96 | |
| 69 | 100 | 100 | 96 | |
| 70 | 100 | 100 | 100 | |
| 71 | 100 | 100 | 100 | |
| 72 | 100 | 100 | 92 | |
| 73 | 100 | 100 | 42 | |
| 74 | 97 | 81 | 42 | |
| 75 | 50 | 49 | 47 | |
As can be seen, the catalyst still retained the activity after calcination. Nevertheless it cannot be used beyond seven additional times; in the eighth cycle both conversion and yield drastically decrease. To see whether or not the catalyst was permanently deactivated it was recalcined again and, after this, it could be used up to six more times with good results but again a drastic reduction of both conversion and yield was observed in the seventh cycle. A third calcination allowed using the recovered catalyst for 10 additional runs with excellent conversions and yields. Additional Pd leaching was determined by ICP-MS analysis of a catalyst sample used in 75 runs, which resulted to be ca. 22% (Table 2, entry 6), in line with the previously found. The reactivation by calcination after complete deactivation points to the catalytic centers are reversibly poisoned by some organic compound which is eliminated by calcination, a behavior not observed in the original catalyst probably due to the protecting role of PVP. It is important to note, however, that PVP is not intrinsically necessary for the catalytic activity of the PNP, since all of it disappears in the first calcination. The stabilizing role of PVP is only key during the early catalyst preparation stages, when PNP are in a colloidal suspension, to control the final particle size and keep the colloidal suspension stable for a long time. Once the PNP are supported on the LAPONITE®, PVP come to have only a secondary role, so it could be eliminated without great consequences. To test this hypothesis, we calcined a sample of freshly prepared catalyst, prior its use in the benchmark reaction (Table 4). As anticipated, the catalyst displayed good activity and recoverability, and could be used in six consecutive runs without apparent loss of activity.
| Run | Conversionc (%) | Yieldc (%) | Isolated yield (%) |
|---|---|---|---|
| a Using a 1% Pd total molar ratio, at 100 °C during 4 hours.b Catalyst calcined at 550 °C under air stream for 10 h prior first use.c Determined by GC using n-decane as the internal standard. Regio- and stereoselectivity to butyl (E)-cinnamate was 100% in all cases. | |||
| 1 | 100 | 100 | 98 |
| 2 | 99 | 99 | 97 |
| 3 | 100 | 100 | 87 |
| 4 | 100 | 100 | 96 |
| 5 | 100 | 100 | 93 |
| 6 | 100 | 100 | 93 |
Catalytic tests in the reactions with styrenes
To enlarge the scope of applicability of this catalyst, and test its stability with different Heck–Mizoroki reactions, we used the PNP-LAPONITE® catalyst in the reaction of iodobenzene with other alkenes: styrene, p-chloro- and p-methoxystyrene (Scheme 2 and Table 5). | ||
| Scheme 2 Heck–Mizoroki reactions between iodobenzene and styrenes, catalyzed by LAPONITE®-supported palladium nanoparticles in solventless conditions. | ||
| R | Time (h) | Run | Conversionb (%) | Yieldb (%) | Isolated yield (%) |
|---|---|---|---|---|---|
| a Using a 1% Pd molar ratio, at 100 °C.b Determined by GC using n-decane as the internal standard. Stereoselectivity to the (E)-stilbene was 100% in all cases.c Regioselectivity to sylbene 90%.d Regioselectivity to sylbene 95%.e Regioselectivity to sylbene 90%. | |||||
| Hc | 8 | 1 | 61 | 55 | 48 |
| 2 | 100 | 100 | 88 | ||
| 3 | 100 | 99 | 88 | ||
| 4 | 45 | 41 | 43 | ||
| 5 | 71 | 67 | 68 | ||
| 6 | 98 | 98 | 95 | ||
| 7 | 100 | 100 | 72 | ||
| 8 | 100 | 100 | 96 | ||
| 9 | 73 | 71 | 74 | ||
| 10 | 99 | 97 | 17 | ||
| 11 | 84 | 79 | 64 | ||
| 12 | 70 | 67 | 70 | ||
| 13 | 60 | 59 | 60 | ||
| 14 | 64 | 62 | 64 | ||
| 15 | 24 | 25 | 25 | ||
| 16 | 64 | 60 | 58 | ||
| 17 | 56 | 47 | 43 | ||
| 18 | 16 | 15 | 17 | ||
| 20 | 1 | 100 | 100 | 66 | |
| 2 | 100 | 100 | 91 | ||
| 3 | 98 | 98 | 97 | ||
| 4 | 100 | 100 | 99 | ||
| 5 | 99 | 98 | 99 | ||
| 6 | 99 | 98 | 89 | ||
| 7 | 100 | 100 | 100 | ||
| 8 | 98 | 98 | 100 | ||
| 9 | 91 | 89 | 82 | ||
| 10 | 91 | 88 | 71 | ||
| 11 | 97 | 97 | 100 | ||
| 12 | 34 | 33 | 33 | ||
| Cld | 20 | 1 | 75 | 73 | 63 |
| 2 | 100 | 100 | 87 | ||
| 3 | 100 | 100 | 89 | ||
| 4 | 100 | 100 | 91 | ||
| 5 | 100 | 100 | 92 | ||
| 6 | 100 | 100 | 91 | ||
| 7 | 100 | 100 | 91 | ||
| 8 | 100 | 100 | 90 | ||
| 9 | 100 | 100 | 96 | ||
| 10 | 100 | 100 | 93 | ||
| 11 | 95 | 95 | 89 | ||
| 12 | 100 | 100 | 93 | ||
| 13 | 95 | 95 | 89 | ||
| 14 | 100 | 100 | 92 | ||
| 15 | 100 | 100 | 95 | ||
| 16 | 100 | 100 | 79 | ||
| 17 | 100 | 100 | 100 | ||
| 18 | 98 | 98 | 91 | ||
| 19 | 92 | 92 | 92 | ||
| 20 | 44 | 40 | 40 | ||
| 21 | 17 | 17 | 17 | ||
| 22 | 70 | 66 | 56 | ||
| MeOe | 20 | 1 | 84 | 83 | 75 |
| 2 | 95 | 95 | 95 | ||
| 3 | 85 | 85 | 77 | ||
| 4 | 100 | 100 | 90 | ||
| 5 | 89 | 89 | 78 | ||
| 6 | 100 | 100 | 83 | ||
| 7 | 72 | 71 | 63 | ||
| 8 | 91 | 91 | 85 | ||
| 9 | 85 | 85 | 78 | ||
| 10 | 72 | 72 | 65 | ||
| 11 | 86 | 85 | 82 | ||
| 12 | 93 | 93 | 84 | ||
| 13 | 44 | 36 | 29 | ||
| 14 | 41 | 40 | 35 | ||
| 15 | 50 | 52 | 49 | ||
| 16 | 95 | 95 | 85 | ||
| 17 | 69 | 64 | 51 | ||
| 18 | 36 | 17 | 12 | ||
As can be seen LAPONITE®-supported nanoparticles are active catalysts in the three reactions tested, although high yields are usually obtained only after eight hours of reaction. Comparison between different substrates have therefore been done using the results after 20 hours of reaction time, because their different reactivity. Some odd results are found in some intermediate cycles (for instance, runs 4 and 5 in the 8 h reactions with styrene, runs 20 and 21 in the reactions with p-chlorostyrene, or runs 13–15 in the reactions with p-methoxystyrene), but these results are related to the aforementioned problems of product extraction, leading to unexpectedly low yields followed by an important increase in the next cycle. Selectivity is not affected by catalyst reuse, so stereoselectivity to the (E)-stilbene is complete in all cases, whereas regioselectivity depends on the nature of the styrene, being about 90% with styrene and p-methoxystyrene, and 95% with p-chlorostyrene. The remaining product is always the corresponding 1,1-diarylethene. Regarding catalyst reuse, it is efficient during 11 cycles with styrene, 12 with p-methoxystyrene and 19 with p-chlorostyrene. The elementary analyses of the used catalysts show again the increase in weight and in carbon and nitrogen contents due to the adsorption of the concomitant ammonium salt on the solid.
The lower stability of the catalyst when used in coupling reactions with styrene had also been observed in previous studies.28 Accounting for the differences in catalyst stability as a function of the nature of the alkene is not an easy task, but it must be noted that alkenes with lower electronic density allow using the catalyst for a higher number of cycles. The comparison of p-chlorostyrene with the other styrenes points towards this behavior, as does the much higher stability of the catalyst when the substrate is an acrylate. At least part of these results can be explained by the much higher Pd leaching observed when styrenes are used as substrates. Fig. 5 displays the linear relationship found between the accumulated yield and the total Pd leaching at the end of a series of consecutive catalytic runs (50, 12, 22 and 18, respectively, for reactions of butyl acrylate, styrene, p-chlorostyrene and p-methoxy-styrene).
 | ||
| Fig. 5 Correlation between the total productivity of the supported PNP catalyst in a series of consecutive reaction runs and the total Pd leaching, as a function of the alkene used in the Heck–Mizoroki reaction with iodobenzene. | ||
It is worth noting that total Pd leaching in the case of the reactions with styrenes is always over 40%, even if the catalysts have been used in less than a half cycles than that of the reaction with butyl acrylate. It seems that Pd leaching, accounting for the differences observed, increases with the electron-donor character of reagents and/or products.
Conclusions
To sum up, the LAPONITE®-supported palladium nanoparticles are efficient catalysts for Heck coupling of iodobenzene with some alkenes. This catalyst can be prepared in a straightforward way, it is easy to handle and use, and shows a high stability and robustness in reaction conditions. Furthermore, the catalyst can be easily recovered and reused, and, in the case of the reaction of butyl acrylate, the catalyst displays an unprecedented recoverability, the highest described to date for any coupling reactions and, in particular, for the Heck–Mirozoki reaction (at least 75 uses and yet without reaching complete deactivation). The delaminated structure and ionic character of the support seem to favor this behavior. Other additional advantages of this catalytic system are that the reaction is carried out in the absence of any additional solvent, allowing the easy separation of the products in pure form because the concomitant ionic byproduct (triethylammonium iodide) remains on the solid together with the catalyst during the product extraction procedure. Finally, the elimination of this byproduct by calcination under air allows reactivation and new reuses of the catalyst.Experimental
General methods
All reagents were purchased from commercial sources (Sigma-Aldrich) and used without further purification, unless otherwise indicated. Deuterated solvents were purchased from Aldrich. All analytical pure reagents were used as the only solvents in the reactions. LAPONITE® clay was obtained from Rockwood Additives Ltd. Conversions and reaction yields were determined by gas chromatography using Agilent 6809A and 7890A gas chromatographs, provided with FID detectors. Compounds were characterized by NMR spectrometry on a Bruker Advance 400 MHz spectrometer. TEM micrographs were obtained with a JEOL-2000 FXII microscope. Field Emission SEM (FESEM) micrographs were obtained with a Carl Zeiss MERLIN™ microscope. Thermogravimetric analyses were carried out in a SDT-2960 Simultaneous TDA-TGA.Preparation of palladium nanoparticles stabilized with polyvinylpyrrolidone (Pd–PVP NPs)
This nanoparticles synthesis method is based on the reduction of a palladium(II) complex with ethanol.30,31 In a 100 mL flask, a 2 mM H2PdCl4 solution was prepared by mixing 35.4 mg of PdCl2 (0.2 mmol), 2 mL of 0.2 M HCl, and 98 mL of H2O. A mixture of 140 mL of H2O, 94 mL of ethanol, 444.6 mg of PVP and 100 mL of the 2 mM H2PdCl4 solution was refluxed in a 500 mL flask during 3 h under air to synthesize the palladium nanoparticles protected by PVP. The black solution was evaporated at reduced pressure, and the remaining black solid was dissolved in 66 mL of ethanol. The result was a black stable colloidal suspension of Pd–PVP NPs (3 mM) in ethanol.Preparation of LAPONITE®-supported palladium nanoparticles
In a 20 mL Schlenk flask, 1 g of LAPONITE®, 1 mL of the ethanol colloidal suspension of Pd nanoparticles (3 mM) and 6 mL of dichloromethane (DCM) were mixed, and the mixture magnetically stirred during 20 min. After that, ethanol and DCM were removed under reduced pressure. The result was a powdered grey solid consisting of the Pd NPs supported onto the LAPONITE® clay.General procedure for Heck coupling reactions
In a 20 mL Schlenk flask containing the solid catalyst, 0.3 mmol of iodobenzene were mixed with 0.7 mmol of the olefin and 2.2 mmol of triethylamine. The reaction mixture got completely adsorbed onto the solid. The Schlenk flask was heated in an oil bath, at 100 °C for 4 h or 20 h, depending on the alkene. After this time, the solid was extracted with hexane at 65 °C (1 × 10 mL and then 4 × 5 mL). The solid was dried under reduced pressure and kept in the same Schlenk flask, ready to be reused, and the combined extracts containing the reaction products were analysed by gas chromatography. Then the solution was filtered through silica, the extraction solvent was removed under reduced pressure, and the isolated product analysed by NMR to test its purity.13C-NMR(100 MHz, CDCl3, APT): 167.1 (−, Cquat), 144.5 (+, CH), 134.5 (−, Cquat), 130.2 (+, CH), 128.9 (+, CH), 128.0 (+, CH), 118.4 (+, CH), 64.4 (−, CH2), 30.8 (−, CH2), 19.2 (−, CH2), 13.7 (+, CH3).
13C-NMR(100 MHz, CDCl3, APT): 137.4 (−, Cquat), 128.7 (+, CH), 128.6 (+, CH), 127.6 (+, CH), 126.5 (+, CH).
13C-NMR(100 MHz, CDCl3, APT): 159.4 (−, Cquat), 137.8 (−, Cquat), 130.2 (−, Cquat), 128.7 (+, CH), 128.3 (+, CH), 127.8 (+, CH), 127.3 (+, CH), 126.7 (+, CH), 126.4 (+, CH), 114.2 (+, CH), 53.4 (+, CH3).
13C-NMR(100 MHz, CDCl3, APT): 137.0 (−, Cquat), 135.9 (−, Cquat), 133.2 (−, Cquat), 129.4 (+, CH), 128.8 (+, CH), 128.7 (+, CH), 127.9 (+, CH), 127.6 (+, CH), 127.4 (+, CH), 126.6 (+, CH).
Acknowledgements
Financial support from the Spanish MINECO (project CTQ2011-28124-C02-01), the European Social Fund (ESF) and the Gobierno de Aragón (Grupo Consolidado E11) is gratefully acknowledged. We also thank Mª Angeles Laguna and Ana C. Gallego, from the Service of Electron Microscopy of Materials of the University of Zaragoza, for their valuable assistance in the TEM and FESEM experiments, to Prof. Antonio Monzón for his valuable assistance in the BET measurements, and to Dr Alfonso Cornejo and Dr Víctor Martínez-Merino, from UPNA, for their valuable advise and collaboration.References
- F. Alonso, I. P. Beletskaya and M. Yus, Tetrahedron, 2008, 64, 3047–3101 CrossRef CAS.
- A. M. Trzeciak and J. J. Ziółkowski, Coord. Chem. Rev., 2007, 251, 1281–1293 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156–181 CrossRef PubMed.
- Y. Liu, Z. Ma, J. Jia, C. Wang, M. Huang and J. Tao, Appl. Organomet. Chem., 2010, 24, 646–649 CrossRef CAS.
- Q. Zhang, H. Su, J. Luo and Y. Wei, Tetrahedron, 2013, 69, 447–454 CrossRef CAS.
- M. Semler, J. Čejka and P. Štěpnička, Catal. Today, 2014, 227, 207–214 CrossRef CAS.
- L. Chen, S. Rangan, J. Li, H. Jiang and Y. Li, Green Chem., 2014, 16, 3978–3985 RSC.
- J.-N. Young, T.-C. Chang, S.-C. Tsai, L. Yang and S. J. Yu, J. Catal., 2010, 272, 253–261 CrossRef CAS.
- L. Yuan, Y. Xu, X. Hu, G. Yang and Y. Wu, J. Mol. Catal. A: Chem., 2015, 396, 55–60 CrossRef CAS.
- A. Biffis, M. Zecca and M. Basato, J. Mol. Catal. A: Chem., 2001, 173, 249–274 CrossRef CAS.
- S. Ogasawara and S. Kato, J. Am. Chem. Soc., 2010, 132, 4608–4613 CrossRef CAS PubMed.
- Q. Du, W. Zhang, H. Ma, J. Zheng, B. Zhou and Y. Li, Tetrahedron, 2012, 68, 3577–3584 CrossRef CAS.
- S. Moussa, A. R. Siamaki, B. F. Gupton and M. S. El-Shall, ACS Catal., 2012, 2, 145–154 CrossRef CAS.
- A. R. Siamaki, Y. Lin, K. Woodberry, J. W. Connell and B. F. Gupton, J. Mater. Chem. A, 2013, 1, 12909–12918 CAS.
- A. R. Siamaki, A. E. R. S. Khder, V. Abdelsayed, M. S. El-Shall and B. F. Gupton, J. Catal., 2011, 279, 1–11 CrossRef CAS.
- E. J. García-Suárez, P. Lara, A. B. García, M. Ojeda, R. Luque and K. Philippot, Appl. Catal., A, 2013, 468, 59–67 CrossRef.
- E. Hariprasad and T. P. Radhakrishnan, ACS Catal., 2012, 2, 1179–1186 CrossRef CAS.
- H. Firouzabadi, N. Iranpoor, M. Gholinejad, S. Akbari and N. Jeddi, RSC Adv., 2014, 4, 17060–17070 RSC.
- G. M. Neelgund and A. Oki, Appl. Catal., A, 2011, 399, 154–160 CrossRef CAS PubMed.
- A. Khazaei, S. Rahmati, Z. Hekmatian and S. Saeednia, J. Mol. Catal. A: Chem., 2013, 372, 160–166 CrossRef CAS.
- B. M. Choudary, S. Madhi, N. S. Chowdari, M. L. Kantam and B. Sreedhar, J. Am. Chem. Soc., 2002, 124, 14127–14136 CrossRef CAS PubMed.
- Z. Zhang and Z. Wang, J. Org. Chem., 2006, 71, 7485–7487 CrossRef CAS PubMed.
- A. V. Martínez, J. A. Mayoral and J. I. García, Appl. Catal., A, 2014, 472, 21–28 CrossRef.
- A. V. Martínez, F. Invernizzi, A. Leal-Duaso, J. A. Mayoral and J. I. García, RSC Adv., 2015, 5, 10102–10109 RSC.
- T. Teranishi and M. Miyake, Chem. Mater., 1998, 10, 594–600 CrossRef CAS.
- Y. Li, E. Boone and M. A. El-Sayed, Langmuir, 2002, 18, 4921–4925 CrossRef CAS.
- K. Köhler, W. Kleist and S. S. Pröckl, Inorg. Chem., 2007, 46, 1876–1883 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Typical GC conditions, NMR characterization of reaction products, catalysts characterization and tables with complete recycling results. See DOI: 10.1039/c5ra10191c |
| This journal is © The Royal Society of Chemistry 2015 |