DOI:
10.1039/C5RA10162J
(Paper)
RSC Adv., 2015,
5, 69479-69486
Highly effective oxygen reduction activity and durability of antimony-doped tin oxide modified PtPd/C electrocatalysts
Received
29th May 2015
, Accepted 7th August 2015
First published on 7th August 2015
Abstract
Antimony-doped tin oxide (ATO) nanoparticles are synthesized by a simple one-step hydrothermal method. The oxygen reduction reaction (ORR) activity of the PtPd/C catalyst is promoted by the presence of ATO. Moreover, after accelerated durability testing, the PtPd/C-ATO catalyst reserves most of its electrochemically active surface area (ESA) and ORR activity compared to the PtPd/C catalyst. The improved electrochemical stability and activity of PtPd/C-ATO is attributed to the high stability of ATO support and the strong interaction between Pt and ATO.
Introduction
Although proton exchange membrane fuel cells (PEMFCs) are ideal future power sources due to their high efficiency, zero emission, low-temperature operation, and fast response,1 there are still some critical issues that need to be overcome before the commercialization of PEMFCs, which include the sluggish kinetics of the oxygen reduction reaction (ORR) and the unsatisfactory long-term durability of the cathode catalysts.2,3 At present, the most widely used cathode catalyst system is Pt in the form of small nanoparticles supported on amorphous carbon particles. These carbon support materials can be oxidized when both oxygen and liquid water are present at high electrode potentials,4,5 and when fuel hydrogen starvation occurs during fuel cell operation.6–8 The electrochemical corrosion of the carbon support causes agglomeration and sintering of the Pt catalyst particles, resulting in a decreased electrochemical surface area (ESA) and deteriorative activity of the catalyst.9 These effects would lead to a rapid degradation of the Pt catalyst and thus shorten the lifetime of the PEMFCs. Consequently, more robust catalysts with enhanced activity and stability, such as Pt-based alloy and core–shell catalysts have been studied as potential alternatives for PEMFCs.10,11
Carbon-supported PtPd catalysts with highly catalytic activity and long-term durability have received increasing interest for the application in ORR. The incorporation of Pd into Pt can modify its electronic state and reduce the local O coverage at high potentials and thus stabilize the Pt surface due to separation and dilution of the active site.12 To solve the carbon corrosion issue, alternative supports are being developed with the objectives to increase both the support durability and the catalyst activity through improving the catalyst-support interaction by replacing6,7,13–17 or combining18,19 carbon with transition metal oxides, such as antimony-doped tin oxide (ATO).20–27 Dou et al.20 used ATO as catalyst support material for oxygen reduction reaction, and found that the ATO support maintains significantly its stability and the performance of the tested electrocatalyst compared to Vulcan XC-72. Furthermore, Yin et al.25 proved that the electrochemical activity and stability of Pt for Pt/ATO/C catalysts is increased with the addition of ATO because of modified electronic structure of Pt by the presence of the ATO phase in the catalyst supports, which was found to significantly enhance the catalyst durability.
In this work, as an effort to improve the durability and activity of the catalysts, we developed a carbon–ATO composite support (abbreviated as C–ATO), and deposited PtPd catalyst on this support for oxygen reduction reaction. For comparison, PtPd supported on pure carbon support was also prepared. Their electrochemical performance and durability were evaluated, and the results are reported here.
Experimental
Material synthesis
The ATO nanoparticles were synthesized by a simple one-step hydrothermal method. Typically, 2 g tin (purity 99.99%) and calculated amount of Sb2O3 (purity 99.99%) (5 at% to Sn) were dissolved in 80 mL 8.3 mol L−1 HNO3 solution and formed a yellow colloid. The colloid solution was transferred to an autoclave and kept at 150 °C for 10 h in an oven. When air cooled to room temperature, the resulting bluish-colored products (characteristic of ATO particle) were collected and washed with water and ethanol, and finally dried at 100 °C for 5 h in an oven. The C–ATO composite supports were prepared by mixing 13.6 mg ATO with 27.2 mg XC-72 in ethanol to obtain well-blended support suspensions.
The PtPd/C–ATO catalyst was prepared by a modified aqueous-phase synthesis method using amphiphilic triblock copolymers as the reductant and capping agent, according to Zhang et al.11 Typically, a given amount of Na2PdCl4 (Aladdin, Shanghai, China) aqueous solution was injected into 14 g L−1 Pluronic F127 (Sigma-Aldrich) solution to give a final PdCl42− concentration of 4.2 mmol L−1. After stirring at 80 °C for 2 h in an oil bath, the Pd colloid solution was obtained. Then calculated amount of Na2PtCl4 (Aladdin, Shanghai, China) aqueous solution (Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pt atom ratio of 2:1) was injected into the above solution, and kept at 80 °C for another 3 h to obtain PtPd nanocrystals (NCs). Afterwards, the as-prepared C–ATO support suspensions were added to the obtained PtPd NCs solution, then the mixture was stirred overnight. The product was collected by centrifugation and dried at 60 °C under vacuum overnight. For comparison, PtPd/C was also prepared by the same method with 40.8 mg XC-72 as support materials. The Pt/C and Pt/C–ATO were synthesized by removing the process of the Pd colloid formation from the preparation of PtPd/C and PtPd/C–ATO, respectively.
:Pt atom ratio of 2:1) was injected into the above solution, and kept at 80 °C for another 3 h to obtain PtPd nanocrystals (NCs). Afterwards, the as-prepared C–ATO support suspensions were added to the obtained PtPd NCs solution, then the mixture was stirred overnight. The product was collected by centrifugation and dried at 60 °C under vacuum overnight. For comparison, PtPd/C was also prepared by the same method with 40.8 mg XC-72 as support materials. The Pt/C and Pt/C–ATO were synthesized by removing the process of the Pd colloid formation from the preparation of PtPd/C and PtPd/C–ATO, respectively.
Material characterizations
X-ray diffraction (XRD) measurements were carried out using a Cu Kα source (PANalytical X'Pert PRO X-ray diffractometer) operated at 40 kV and 40 mA. Transmission electron microscope (TEM) characterization was performed on a JEOL JEM-2000EX microscope. The Brunauer–Emmet–Teller (BET) area was estimated using a QuadraSorb SI4 system. Elemental analysis was carried out on a JEOL 6360LV scanning electron microscopy equipped with an energy dispersive X-ray spectrometer (EDX). The XPS spectra were obtained on an ESCALAB250XI spectrometer and the binding energies were calibrated according to the C 1s peak (284.8 eV). The conductivities of support materials were tested using four-point probe measuring system (Suzhou Jingge Electronic Co., China).
Electrochemical measurements
All electrochemical measurements were conducted using a CHI730 electrochemical station. Pt foil and saturated calomel electrode (SCE) were employed as the counter and reference electrode, respectively. All the potentials are given versus the normal hydrogen electrode (NHE). Working electrode was prepared by coating appropriate amount of electrocatalyst and Nafion® on the glassy carbon electrode (d = 4 mm) according to the literature.28,29 Catalyst ink was obtained by sonicating 5 mg of catalyst, 50 μL Nafion® solution (5 wt%, Alfa Aesar), and 1 mL isopropanol into homogeneous slurry. Then, 4 μL this ink was dropped on the glassy carbon electrode and allowed to dry in air at room temperature. All cyclic voltammetry (CV) measurements were profiled in 0.5 mol L−1 H2SO4 solution deaerated with high purity N2 in the potential range of 0.02 and 1.2 V at a scan rate of 50 mV s−1. The oxygen reduction curve was measured in oxygen saturated 0.5 mol L−1 H2SO4 from 1.0 to 0.2 V at 10 mV s−1 with a rotating speed of 1600 rpm. A potential cycling test from 0.6 V to 1.2 V were conducted to examine the electrochemical stability of the catalysts. The electrochemical surface area (ESA) of catalysts was estimated according to the charge of hydrogen desorption after double-layer correction, assuming monolayer hydrogen adsorption on Pt surface (0.21 mC cm−2).
Results and discussion
Characterization of ATO support material
Fig. 1a and b show TEM image and corresponding particle size distribution histogram of ATO. As seen from the TEM image, the ATO support material is composed of nanoparticles and has a relatively narrow particle size distribution. On the basis of measuring the size of more than 100 randomly chosen particles in the TEM image, the mean particle diameter of ATO support is 3.3 nm. Energy dispersive X-ray spectroscopy (EDX) analysis of the as-prepared support proved the presence of Sb and Sn in 5.4:100 atomic ratio for ATO, which agrees closely with the initial atomic ratio (5.0:100) used in ATO preparation.
 |
| | Fig. 1 (a) TEM image and (b) corresponding particle size distribution histogram of ATO. | |
Fig. 2 displays the XRD pattern of the ATO support material, all of the diffraction positions and relative intensities of support material match well with standard XRD pattern of cassiterite SnO2 (JCPDS card no. 00-021-1250). The main characteristic diffraction patterns at 2θ = 26.5, 33.8, 37.9, and 51.7, correspond to the planes of (110), (101), (220), and (211), respectively. Moreover, there are no peaks arising from impurity, such as Sb, Sb2O3 and Sb2O5. The average size of the ATO nanoparticles calculated from the (110) peak using Scherrer's equation is 3.5 nm, which is in accordance with the results obtained by TEM. Fig. 3 exhibits the nitrogen adsorption/desorption isotherms of ATO support. As can be observed, the surface area of the ATO support synthesized in this study is found to be 146.5 m2 g−1. In addition, the electronic conductivity of the ATO nanoparticles is measured as approximately 1.47 × 10−3 S cm−1.
 |
| | Fig. 2 XRD pattern of ATO. | |
 |
| | Fig. 3 Nitrogen adsorption/desorption isotherms of ATO. | |
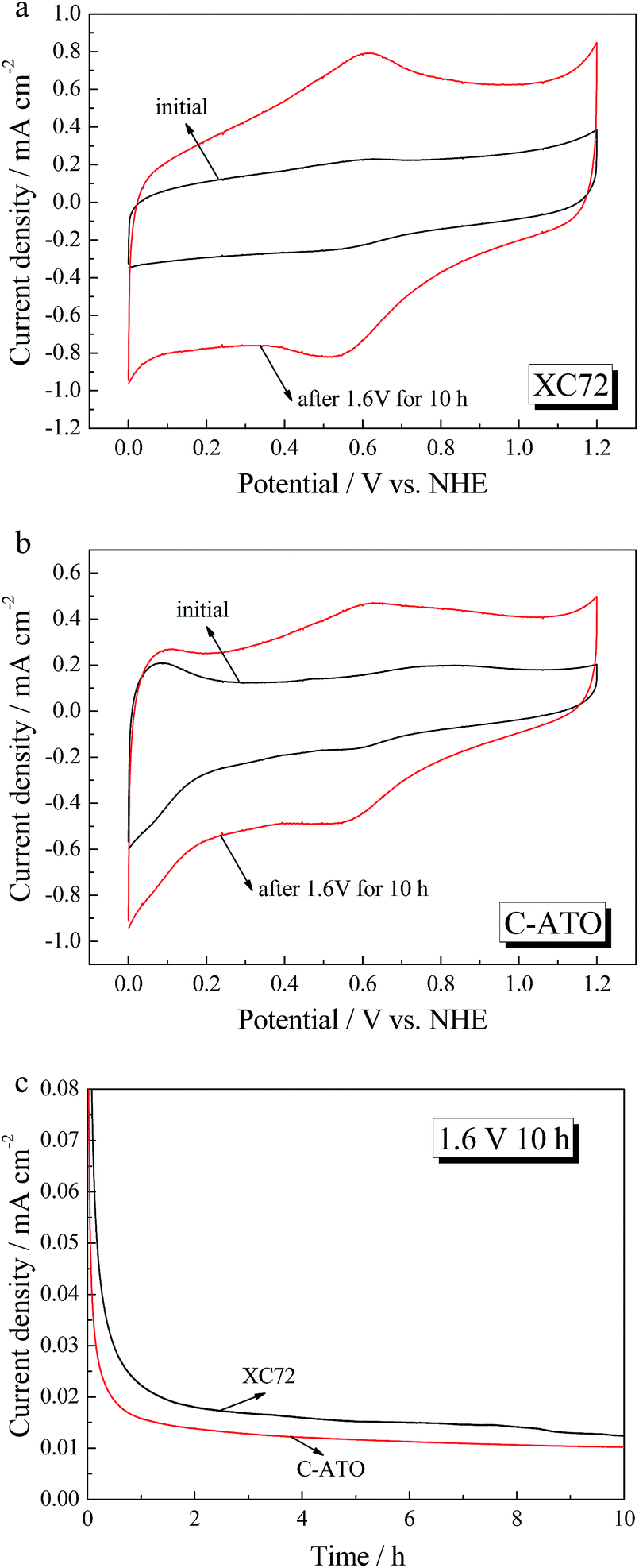
The electrochemical stabilities of XC-72 and C-ATO supports are studied by a rotating disk electrode (RDE) in 0.5 mol L−1 H2SO4 at an elevated potential. Fig. 4a and b show the CV curves of XC-72 and C–ATO before and after potential hold at 1.6 V for 10 h. As seen in Fig. 4a, the carbon support has a significant increase in the oxidation current after potential holding. The large redox couple at 0.6 V indicates that severe carbon corrosion has occurred. For C–ATO, there is still a redox couple at 0.6 V because of the presence of XC-72. However, the corrosion current of C–ATO after 1.6 V for 10 h is smaller than that of XC-72 (see Fig. 4c), indicating that the prepared C–ATO composite support is rather more stable than commercial XC-72 carbon when subjected to high potentials.
 |
| | Fig. 4 CV curves of XC-72 (a) and C-ATO (b) before and after 1.6 V oxidation for 10 h in 0.5 mol L−1 H2SO4 electrolyte with a scan rate of 50 mV s−1; (c) chronoamperometric curves of XC-72 and C–ATO measured at 1.6 V. | |
Characterization of the catalysts
Fig. 5 displays the TEM images of PtPd/C and PtPd/C–ATO samples. For PtPd/C catalyst (Fig. 5a), there are some irregular nanoparticles distributing on the carbon support. However, when ATO is added, the active component is distributing on both of the carbon support and the ATO support in the form of the clusters composed of nanoparticles (see Fig. 5b).
 |
| | Fig. 5 TEM images of PtPd/C (a) and PtPd/C–ATO (b). | |
The XRD patterns of PtPd/C and PtPd/C–ATO are shown in Fig. 6. The diffraction peaks of the PtPd/C catalyst at around 40°, 47°, 68°, 81°, and 86° are attributed to the (111), (200), (220), (311) and (222) planes of the PtPd alloy (JCPDS card no. 03-065-6418), suggesting good crystallinity of these homemade PtPd NCs. The diffraction peak at around 25° comes from the carbon black. In terms of the PtPd/C–ATO catalyst, it can be seen that the catalyst displays characteristic peaks of PtPd crystalline structure, together with diffraction patterns of oxide component (ATO) with rutile SnO2 structure which also can be found in Fig. 2. In addition, the mean crystallite size of PtPd NCs in each catalyst could be calculated by Scherrer's equation and found to be 12 and 11 nm for PtPd/C and PtPd/C–ATO catalysts, respectively.
 |
| | Fig. 6 XRD patterns of PtPd/C and PtPd/C–ATO. | |
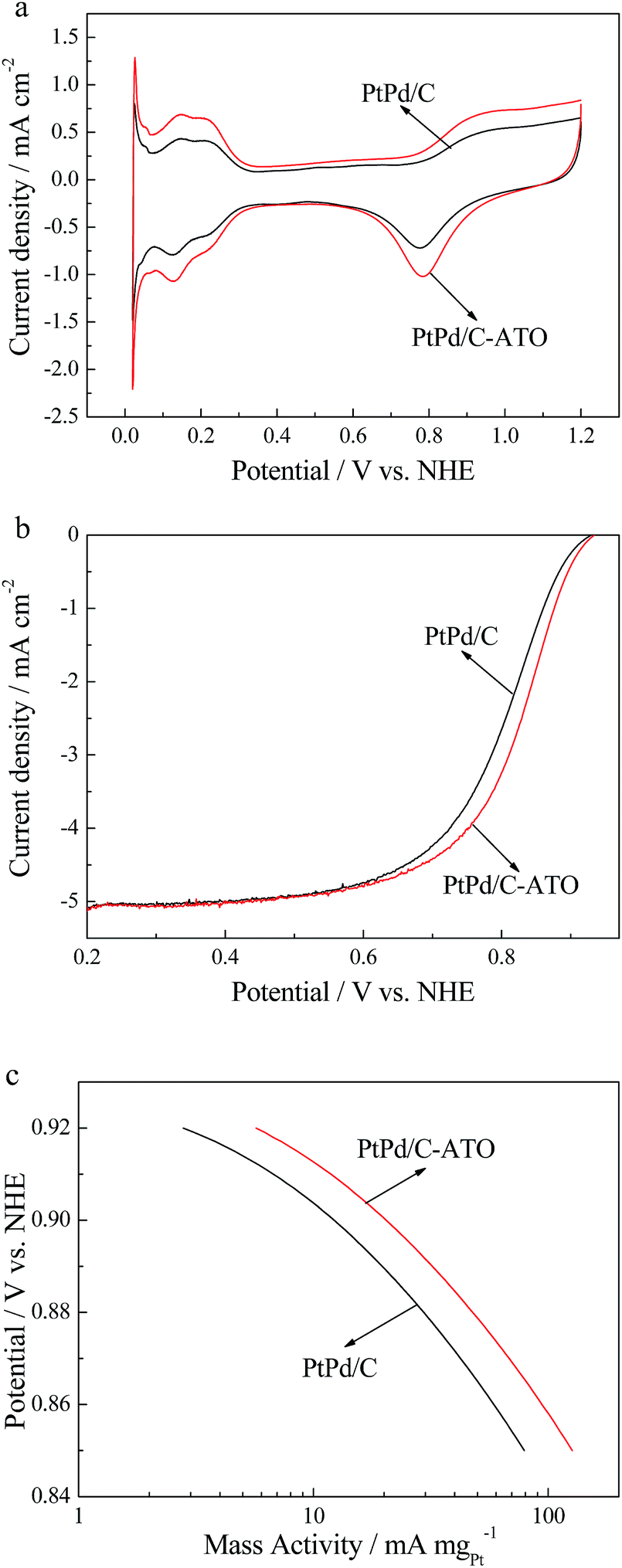
The electrocatalytic performances of PtPd/C and PtPd/C–ATO toward ORR are investigated in 0.5 mol L−1 H2SO4 at room temperature. The CV and ORR polarization curves for different catalysts are presented in Fig. 7a and b. The Pt loading of PtPd/C and PtPd/C–ATO catalysts obtained by EDX are 13.49 wt% and 14.23 wt%, respectively. As seen from Fig. 7a, the sharp peak, appeared between 0.02 and 0.05 V vs. NHE, may be ascribed to hydrogen absorption on Pd. The initial ESA, which is based on the Pt mass of the PtPd/C–ATO catalyst, is estimated to be 40.9 m2 g−1, which is higher than that of the PtPd/C catalyst (27.5 m2 g−1). The ORR polarization curves (Fig. 7b) of the two catalysts show that the PtPd/C–ATO catalyst has more positive onset potential and half-wave potential than PtPd/C, which indicates the higher catalytic activity of PtPd/C–ATO than PtPd/C catalyst. The Pt mass activity (MA) (Fig. 7c) is obtained by normalizing the kinetic current densities ik against the Pt mass of these catalysts. The ORR mass activity of the PtPd/C–ATO catalyst at 0.9 V is 20.4 mA mgPt−1, higher than that of PtPd/C (12.2 mA mgPt−1).
 |
| | Fig. 7 (a) CV curves, (b) ORR polarization curves and (c) MA of PtPd/C and PtPd/C–ATO. | |
In order to understand the reason for an increase in the performance of the ORR activity by introduction of ATO, XPS analysis is conducted. Fig. 8a–d show XPS spectra in Pt4f region for Pt/C, Pt/C–ATO, PtPd/C and PtPd/C–ATO, respectively. The Pt4f peak in Fig. 8 can be decomposed into pairs of three peaks, which are assignable to Pt(0), Pt(II) and Pt(IV) in each of the pairs. The binding energies (BE) of Pt4f7/2 along with the relative densities of Pt(0) for each catalyst are listed in Table 1. As for the Pt/C–ATO catalyst, the BE of the Pt(0) is shifted to the lower energy with the addition of ATO on the support compared to the Pt/C catalyst. This result indicates the modulated interactions between Pt and supports by introduction of the ATO. In addition, the content of Pt(0) is found to be gradually increased from 60.18% for Pt/C to 72.07% for the Pt/C–ATO (see Table 1), further demonstrating the strong metal–support interactions between Pt and ATO. Similar results have also been observed by Yin et al.25 In terms of the PtPd/C and PtPd/C–ATO catalysts, the BE of the Pt(0) for PtPd/C–ATO shifts positively by 0.08 eV compared to PtPd/C. The result is different from the shift of the BE between Pt/C and Pt/C–ATO. From XPS analysis, we can see that the surface Pt/Pd molar ratio of the PtPd/C catalyst is 0.65:1. As for the PtPd/C–ATO, the surface Pt/Pd molar ratio is 0.57:1. That is, when ATO is added, the surface Pt content decreases. Zhang et al.30 suggest that the less Pt content in the surface of the catalyst, the more positive shift in the BE of the Pt in the Co@Pt/C catalysts. Therefore, the BE of the Pt(0) for PtPd/C shifts positively when ATO is added due to the less Pt content in the PtPd/C–ATO catalyst. Moreover, the content of Pt(0) for PtPd/C–ATO is 75.98%, higher than that of the PtPd/C (73.29%) (see Table 1). The result is similar to Liu et al.,31 concluding that the content of Pt(0) in the PtAu/C increases with the addition of TiO2. Thus, the Pt in the PtPd/C–ATO possesses higher activity and stability due to the greater corrosion resistance of Pt(0).25
 |
| | Fig. 8 XPS spectra for Pt4f region of Pt/C (a), Pt/C–ATO (b), PtPd/C (c) and PtPd/C–ATO (d). | |
Table 1 Quantitative data of the fits of Pt 4f7/2 XPS spectra for Pt/C, Pt/C–ATO, PtPd/C and PtPd/C–ATO
| Samples |
BE of Pt(0)/eV |
Values of Pt(0) in % in the total intensity |
| Pt/C |
71.86 |
60.8 |
| Pt/C–ATO |
71.76 |
72.07 |
| PtPd/C |
71.54 |
73.29 |
| PtPd/C–ATO |
71.62 |
75.98 |
Furthermore, the long-term stability of PtPd/C and PtPd/C–ATO catalysts are evaluated by applying potential cycling between 0.6 and 1.2 V in N2-purged 0.5 mol L−1 H2SO4, and the CV curves before and after degradation are shown in Fig. 9a and b. After 1500 cycles, the PtPd/C–ATO catalyst losts 67.5% of its initial Pt ESA, whereas the PtPd/C catalyst losts approximately 82.2% of its initial ESA. In addition, the mass activities of the two catalysts estimated from kinetic current densities versus the cycle number are shown in Fig. 9c. As shown, the Pt mass activities of the PtPd/C and PtPd/C–ATO catalysts are enhanced in the first 100 potential cycles, which may be attributed to the compositional change of PtPd nanoalloy during potential cycling.32 Some researches32,33 have reported that the non-noble metal in Pt-M alloy catalysts can be easily dissolved under electrochemical operating conditions, leaving the surface of the catalyst Pt-rich. Fig. 10 shows XPS spectra of the PtPd/C and PtPd/C–ATO catalysts before and after ADT in the binding energy ranges of Pd3d and Pt4f. It can be seen in Fig. 10b that the Pd peaks of the PtPd/C and PtPd/C–ATO catalysts are decreased significantly, while the peak intensity of Pt (Fig. 10a) reduces more slightly, suggesting remarkable Pd dissolution and little Pt loss after potential cycling. According to the intensity ratio of Pd3d and Pt4f, as derived from the area under the peaks, the Pd/Pt surface atomic ratio of the PtPd/C catalyst before ADT is evaluated to be 1.44:1, while decreases to 0.84:1 after ADT. The results indicate Pt enrichment on the outer layer. Similarly, the Pd/Pt surface atomic ratio of the PtPd/C–ATO catalyst after ADT (0.57:1) is smaller than that of the PtPd/C–ATO catalyst before ADT (1.76:1). The smaller lattice parameter of Pd-rich core would induce compressive strain in the outer shell, which would tend to downshift the d-band center of the Pt-rich shell,34 weakening the adsorption energy of the oxygenated species, and thus enhancing the ORR kinetics of the dealloyed PtPd/C and PtPd/C–ATO catalysts.35 Afterwards, the Pt mass activities for both of the catalysts are reduced during the potential cycling due to the significant decrease of ESA. The ORR activity of the PtPd/C–ATO catalyst at 0.9 V decreases to 54.4% of its initial value, while the PtPd/C catalyst retains only 39.4% of its initial ORR activity after 1500 cycles. Meanwhile, the ORR activity of the PtPd/C–ATO catalyst is 2.3 times higher than that of the PtPd/C catalyst after potential cycling. These results confirm that the PdPt/C–ATO catalyst is more durable than PdPt/C catalyst (Table 2).
 |
| | Fig. 9 CV curves of (a) PtPd/C and (b) PtPd/C–ATO under 1500 potential cycling between 0.6 and 1.2 V; (c) MA given as kinetic current densities normalized against the Pt mass of PtPd/C and PtPd/C–ATO as a function of cycle numbers. | |
 |
| | Fig. 10 XPS spectra of the PtPd/C and PtPd/C–ATO catalysts before and after ADT in the binding energy ranges of (a) Pt4f and (b) Pd3d. | |
Table 2 The mass activity (MA), specific activity (SA), electrochemical surface area (ESA), and decay rate of the PtPd/C and PtPd/C–ATO catalysts before and after potential cycling between 0.6 and 1.2 V vs. NHE at 50 mV s−1 for 1500 cycles in N2-purged 0.5 mol L−1 H2SO4
| Samples |
ESA/m2 g−1 |
MA/mA mg−1 |
SA/A m−2 |
| PtPd/C |
0 |
27.5 |
12.2 |
1.0 |
| 1500 |
4.9 |
4.8 |
0.98 |
| Loss |
82.2% |
60.6% |
2% |
| PtPd/C–ATO |
0 |
40.9 |
20.4 |
0.5 |
| 1500 |
13.3 |
11.1 |
0.83 |
| Loss |
67.5% |
45.6% |
−66% |
Conclusions
ATO nanoparticles with large surface area were successfully synthesized via a simple one-step hydrothermal method. The electrochemical stability of XC-72 improved when ATO was added. Meanwhile, the PtPd/C–ATO catalyst showed significantly enhanced catalytic activity for the ORR compared with the PtPd/C catalyst. The improvement for activity was attributed to the high ESA and modified electronic structure of Pt by the presence of the ATO phase in the catalyst supports which was also found to significantly enhance the catalyst durability.
Acknowledgements
We gratefully acknowledge the financial support of this research by the National High Technology Research and Development Program (No. 2013AA110201) and the Key Project of Natural Science Foundation (No. 201436003).
References
- N. Yousfi-Steiner, P. Mocotéguy, D. Candusso, D. Hissel, A. Hernandez and A. Aslanides, J. Power Sources, 2008, 183, 260 CrossRef CAS PubMed.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K.-I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, Carbon, 2005, 43, 179 CrossRef CAS PubMed.
- Y.-J. Wang, D. P. Wilkinson and J. Zhang, Dalton Trans., 2012, 41, 1187 RSC.
- M. Dou, M. Hou, D. Liang, W. Lu, Z. Shao and B. Yi, Electrochim. Acta, 2013, 92, 468 CrossRef CAS PubMed.
- M. Dou, M. Hou, H. Zhang, G. Li, W. Lu, Z. Wei, Z. Shao and B. Yi, ChemSusChem, 2012, 5, 945 CrossRef CAS PubMed.
- C. V. Subban, Q. Zhou, A. Hu, T. E. Moylan, F. T. Wagner and F. J. DiSalvo, J. Am. Chem. Soc., 2010, 132, 17531 CrossRef CAS PubMed.
- V. T. T. Ho, C.-J. Pan, J. Rick, W.-N. Su and B.-J. Hwang, J. Am. Chem. Soc., 2011, 133, 11716 CrossRef CAS PubMed.
- V. R. Stemencovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef PubMed.
- G. Zhang, Z. Shao, W. Lu, H. Xiao, F. Xie, X. Qin, J. Li, F. Liu and B. Yi, J. Phys. Chem. C, 2013, 117, 13413 CAS.
- H. Li, G. Sun, N. Li, S. Sun, D. Su and Q. Xin, J. Phys. Chem. C, 2007, 111, 5605 CAS.
- C. Yao, F. Li, X. Li and D. Xia, J. Mater. Chem., 2012, 22, 16560 RSC.
- C. Zhang, H. Yu, Y. Li, W. Song, B. Yi and Z. Shao, Electrochim. Acta, 2012, 80, 1 CrossRef CAS PubMed.
- Y. Liu and W. E. Mustain, J. Am. Chem. Soc., 2012, 135, 530–533 CrossRef PubMed.
- S. Huang, P. Ganesan and B. N. Popov, Appl. Catal., B, 2011, 102, 71 CrossRef CAS PubMed.
- Y. Gao, M. Hou, Z. Shao, C. Zhang, X. Qin and B. Yi, J. Energy Chem., 2014, 23, 331 CrossRef CAS.
- B. Ruiz-Camacho, J. H. Martínez-González, R. G. González-Huerta and M. Tufiño-Velázquez, Int. J. Hydrogen Energy, 2014, 39, 16731 CrossRef CAS PubMed.
- N. Zhang, S. Zhang, C. Du, Z. Wang, Y. Shao, F. Kong, Y. Lin and G. Yin, Electrochim. Acta, 2014, 117, 413 CrossRef CAS PubMed.
- M. Dou, M. Hou, F. Wang, D. Liang, Q. Zhao, Z. Shao and B. Yi, J. Electrochem. Soc., 2014, 161, F1231 CrossRef PubMed.
- K.-S. Lee, I.-S. Park, Y.-H. Cho, D.-S. Jung, N. Jung, H.-Y. Park and Y.-E. Sung, J. Catal., 2008, 258, 143 CrossRef CAS PubMed.
- M. P. Gurrola, M. Guerra-Balcázar, L. Álvarez-Contreras, R. Nava, J. Ledesma-García and L. G. Arriaga, J. Power Sources, 2013, 243, 826 CrossRef CAS PubMed.
- K. Kakinuma, Y. Chinoa, Y. Senoo, M. Uchida, T. Kamino, H. Uchid, S. Deki and M. Watanabe, Electrochim. Acta, 2013, 110, 316 CrossRef CAS PubMed.
- F. Takasaki, S. Matsuie, Y. Takabatake, Z. Noda, A. Hayashi, Y. Shiratori, K. Ito and K. Sasaki, J. Electrochem. Soc., 2011, 158, B1270 CrossRef CAS PubMed.
- M. Yin, J. Xu, Q. Li, J. O. Jensen, Y. Huang, L. N. Cleemann, N. J. Bjerrum and W. Xing, Appl. Catal., B, 2014, 144, 112 CrossRef CAS PubMed.
- C. Pan, Y. Li, Y. Ma, X. Zhao and Q. Zhang, J. Power Sources, 2011, 196, 6228 CrossRef CAS PubMed.
- J. Xu, D. Aili, Q. Li, C. Pan, E. Christensen, J. O. Jensen, W. Zhang, G. Liu, X. Wang and N. J. Bjerrum, J. Mater. Chem. A, 2013, 1, 9737 CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS PubMed.
- X. Zhang, H. Wang, J. Key, V. Linkov, S. Ji, X. Wang, Z. Lei and R. Wang, J. Electrochem. Soc., 2012, 159, B270 CrossRef CAS PubMed.
- C. Liu, H. Chen, C. Lai, J. Lin, L. Tsai and K. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 1589 CAS.
- W. He, H. Jiang, Y. Zhou, S. Yang, X. Xue, Z. Zou, X. Zhang, D. L. Akins and H. Yang, Carbon, 2012, 50, 265 CrossRef CAS PubMed.
- J. Zhao and A. Manthiram, Appl. Catal., B, 2011, 101, 660 CrossRef CAS PubMed.
- J. Zhang, Y. Mo, M. B. Vukmirovic, R. Klie, K. Sasaki and R. R. Adzic, J. Phys. Chem. B, 2004, 108, 10955 CrossRef CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.