
Synthesis and characterization of rare-earth metal guanidinates stabilized by amine-bridged bis(phenolate) ligands and their application in the controlled polymerization of rac-lactide and rac-β-butyrolactone†
Tinghua Zeng,
Qinqin Qian,
Bei Zhao*,
Dan Yuan*,
Yingming Yao* and
Qi Shen
Key Laboratory of Organic Synthesis of Jiangsu Province and Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Dushu Lake Campus, Suzhou, 215123, People's Republic of China. E-mail: zhaobei@suda.edu.cn; yuandan@suda.edu.cn; yaoym@suda.edu.cn; Fax: +86-512-65880305; Tel: +86-512-65882806
First published on 9th June 2015
Abstract
Eight rare-earth metal guanidinates supported by a versatile family of chelating amine-bridged bis(phenolate) ligands were synthesized. Metathesis reactions of rare-earth metal chlorides [LnClL1(THF)] stabilized by amine-bridged bis(phenolate) ligand L1 with in situ generated lithium guanidinates in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in THF afforded ytterbium guanidinates YbL1 [R2NC(NR1)2] [R1 = –iPr, R2N = –NiPr2 (1), –N(CH2)5 (2)]. Insertion reactions of the yttrium amides bearing bridged bis(phenolate) ligands with 1 equiv of N,N′-diisopropylcarbodiimide (DIC) yielded six yttrium guanidinates YL1 [(SiHMe2)2NC(NiPr)2] (3), YL2[(SiHMe2)2NC(NiPr)2](THF) (4), YL3[(SiHMe2)2NC(NiPr)2] (5), YL4[(SiHMe2)2NC(NiPr)2] (6), YL5 [(SiHMe2)2NC(NiPr)2] (7), YL6[(SiHMe2)2NC(NiPr)2] (8), respectively. The behaviors of complexes 1–8 in the polymerization of rac-lactide (LA) and rac-β-butyrolactone (BBL) were also explored. It was found that complexes 1–8 efficiently initiated the ring-opening polymerization (ROP) of rac-LA and rac-BBL in a controlled manner, providing highly heterotactic polylactide (Pr up to 0.99) and highly syndiotactic poly(3-hydroxybutyrate) (Pr up to 0.82). The framework of the bridge played a significant role in governing the stereoselectivity, while guanidinate groups work as initiating groups.
:1 molar ratio in THF afforded ytterbium guanidinates YbL1 [R2NC(NR1)2] [R1 = –iPr, R2N = –NiPr2 (1), –N(CH2)5 (2)]. Insertion reactions of the yttrium amides bearing bridged bis(phenolate) ligands with 1 equiv of N,N′-diisopropylcarbodiimide (DIC) yielded six yttrium guanidinates YL1 [(SiHMe2)2NC(NiPr)2] (3), YL2[(SiHMe2)2NC(NiPr)2](THF) (4), YL3[(SiHMe2)2NC(NiPr)2] (5), YL4[(SiHMe2)2NC(NiPr)2] (6), YL5 [(SiHMe2)2NC(NiPr)2] (7), YL6[(SiHMe2)2NC(NiPr)2] (8), respectively. The behaviors of complexes 1–8 in the polymerization of rac-lactide (LA) and rac-β-butyrolactone (BBL) were also explored. It was found that complexes 1–8 efficiently initiated the ring-opening polymerization (ROP) of rac-LA and rac-BBL in a controlled manner, providing highly heterotactic polylactide (Pr up to 0.99) and highly syndiotactic poly(3-hydroxybutyrate) (Pr up to 0.82). The framework of the bridge played a significant role in governing the stereoselectivity, while guanidinate groups work as initiating groups.
Introduction
With the growing global concern over sustainability, the preparation of biodegradable polymers such as poly(lactic acid) (PLA) and poly(3-hydroxybutyrate) (PHB) has become increasingly important and opened commercial prospects for a range of applications in the pharmaceutical, biomedical, and environmental fields.1–6 The most straightforward route to prepare PLA and PHB is the ring-opening polymerization (ROP) of the corresponding cyclic monomers, promoted by initiators that achieve high activity with controlled molecular weight, polydispersity, and microstructure of resulting polymers.1,4,7 Among the variety of metal complexes that have been disclosed for these polymerizations, rare-earth metal complexes have been reported to be efficient initiators for the preparation of unique polyesters with controlled features.8–28Amine bridged bis(phenolate)s, as one type of dianionic chelating ligands, have received considerable attention in rare-earth metal chemistry due to their easy availability, highly tunable features and outstanding capacity to stabilize metal centers. Rare-earth metal complexes stabilized by amine bridged bis(phenolate) ligands have been reported to be efficient initiators for the ROP of cyclic esters, yielding polymers in high yields.9,11–18,21–27,29–32 Recently, our group has reported a series of rare-earth metal complexes supported by bridged bis(phenolate) ligands.21–26,29–36 Lanthanide aryloxides and alkoxides stabilized by amine-bridged bis(phenolate) ligands are found to be efficient initiators for the controlled polymerization of rac-lactide (LA) and rac-β-butyrolactone (BBL).22–25 Moreover, group IV metal complexes bearing an amine-bridged bis(phenolato) ligand are highly efficient in catalyzing regioselective intermolecular hydroamination reactions.37
To gain better understanding of the relationship between the bis(phenolate) ligands and the catalytic property of corresponding rare-earth metal complexes, we have designed and synthesized new guanidinato complexes stabilized by several amine bridged bis(phenolate) ligands. Although guanidinato ligands have already been reported to stabilize rare-earth metal complexes, they were not employed as initiating groups in ROP of cyclic esters.8c,38–40 Complexes bearing both bis(phenolate) and guanidinato ligands are thus tested in initiating the ROP of rac-lactide and rac-β-butyrolactone, and their activities and capacities in stereocontrol are compared.
Results and discussion
Synthesis and characterization of complexes 1–8
Ligand precursors L1H2–L6H2 bearing various bridges were synthesized by Mannich condensations of substituted phenols, formaldehydes, and appropriate amines.41 They were isolated in moderate to good yields as white powders (Chart 1).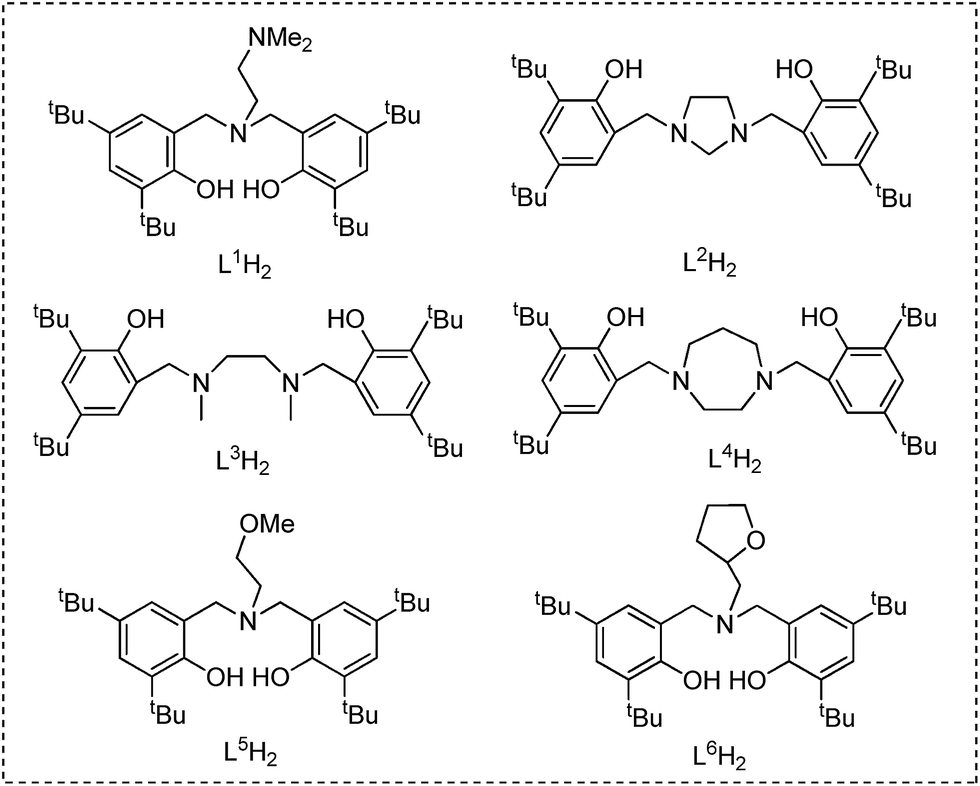 | ||
| Chart 1 Ligand precursors L1H2–L6H2. | ||
Two general methods for synthesizing rare-earth metal guanidinates include metathesis reactions between a suitable rare-earth metal precursor and an alkali guanidinate,38,39 and the insertion of carbodiimides into a rare-earth metal–nitrogen bond.40 Ytterbium guanidinato complexes 1–2 were synthesized from metathesis reactions between the easily available Yb(THF)ClL1 and freshly prepared lithium guanidinates (Scheme 1). Yttrium complexes 3–8 were prepared via insertion reactions. In a two-step sequence, ligand precursors LH2 first reacted with Y[N(SiHMe2)2]3(THF)2 to generate yttrium amides, which were treated with one equivalent of N,N′-diisopropylcarbodiimide (DIC) affording six yttrium guanidinates 3–8 (Scheme 2 and Chart 2).
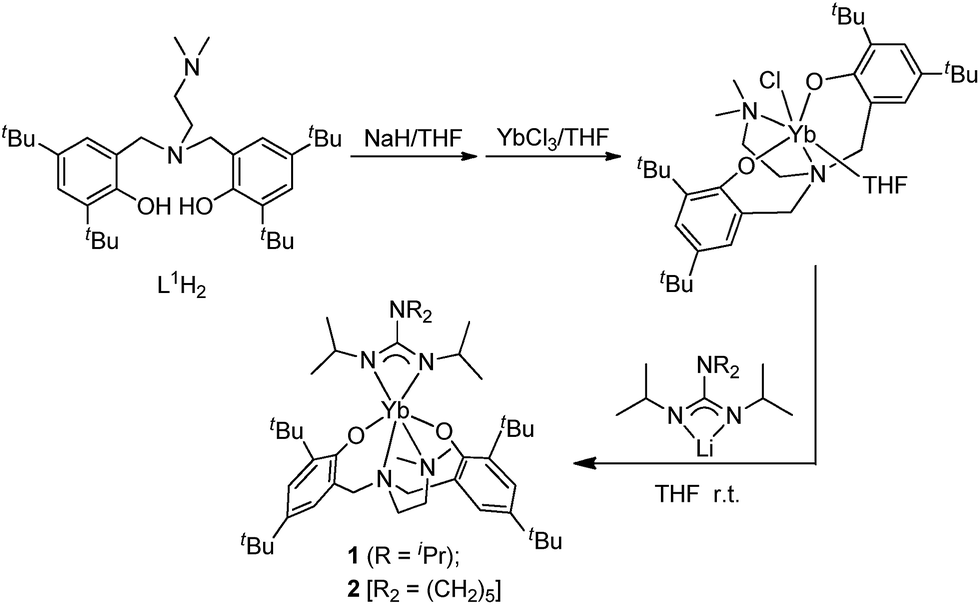 | ||
| Scheme 1 Synthesis of complexes 1–2. | ||
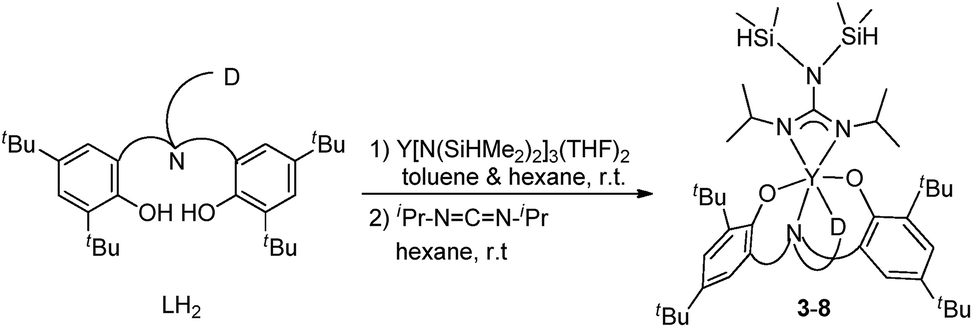 | ||
| Scheme 2 Synthesis of complexes 3–8. | ||
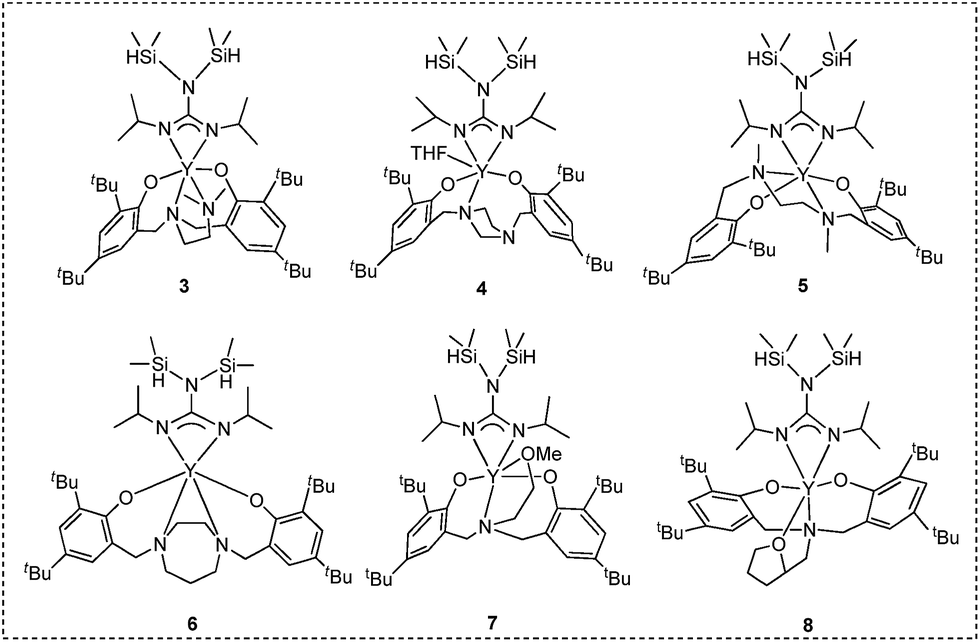 | ||
| Chart 2 Complexes 3–8. | ||
Complexes 1 and 2 are well soluble in THF, moderately soluble in toluene, and slightly soluble in hexane. Complexes 3–8 are moderately soluble in hexane. All complexes are quite air- and moisture-sensitive, but both the crystals and solution are quite stable when stored under argon. These complexes were characterized by elemental analysis, IR spectroscopy, X-ray diffraction studies in the cases of complexes 1, 2, 4 and 5, and by NMR spectroscopy for diamagnetic complexes 3–8.
Crystal structure analyses
Crystals suitable for X-ray diffraction analysis of complexes 1 and 2 were obtained from toluene and THF solution at room temperature, respectively, whereas crystals of complexes 4 and 5 were obtained from hexane solution, respectively. X-ray diffraction analyses show that complexes 1, 2, 4 and 5 have monomeric structures. The ORTEP diagrams of all four structures are depicted in Fig. 1–4, respectively. Their crystallographic data are given in Table 1, and the selected bond lengths and angles are provided in Table 2.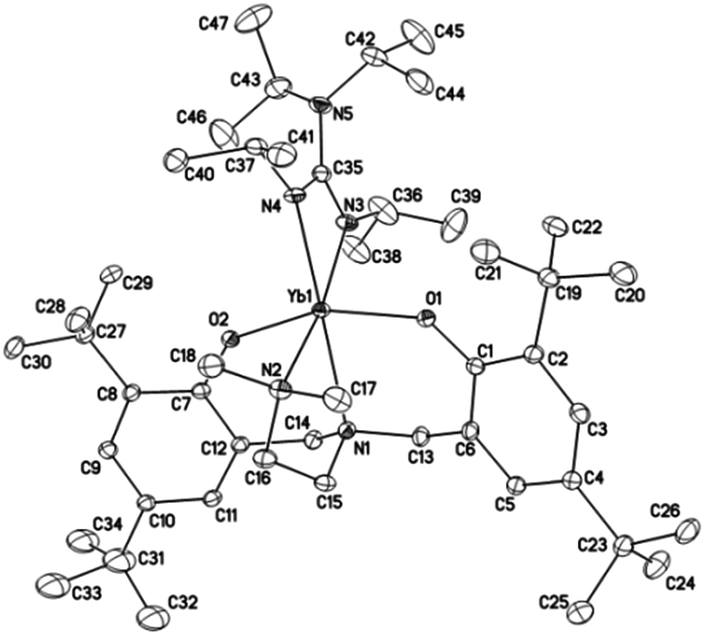 | ||
| Fig. 1 ORTEP diagram of complex 1 showing the atom numbering scheme. Thermal ellipsoids are drawn at the 20% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
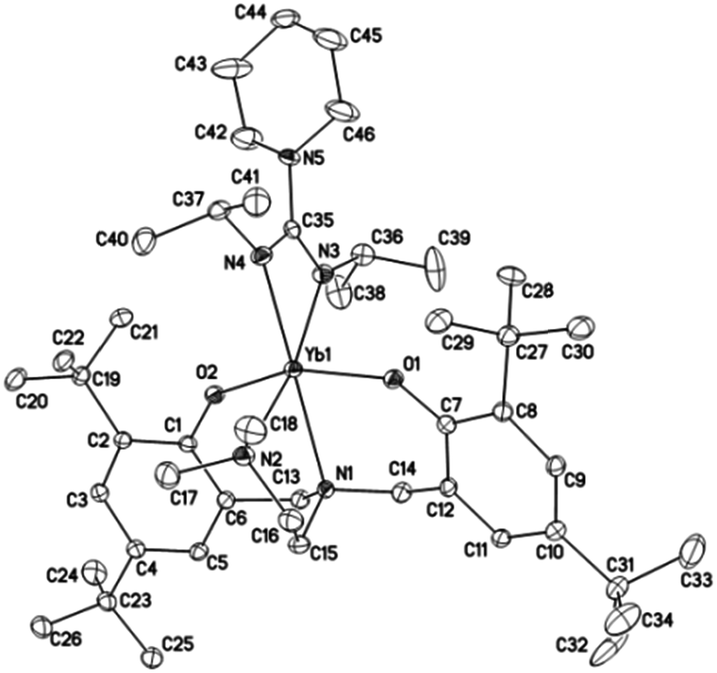 | ||
| Fig. 2 ORTEP diagram of complex 2 showing the atom numbering scheme. Thermal ellipsoids are drawn at the 20% probability level. Hydrogen atoms are omitted for clarity. | ||
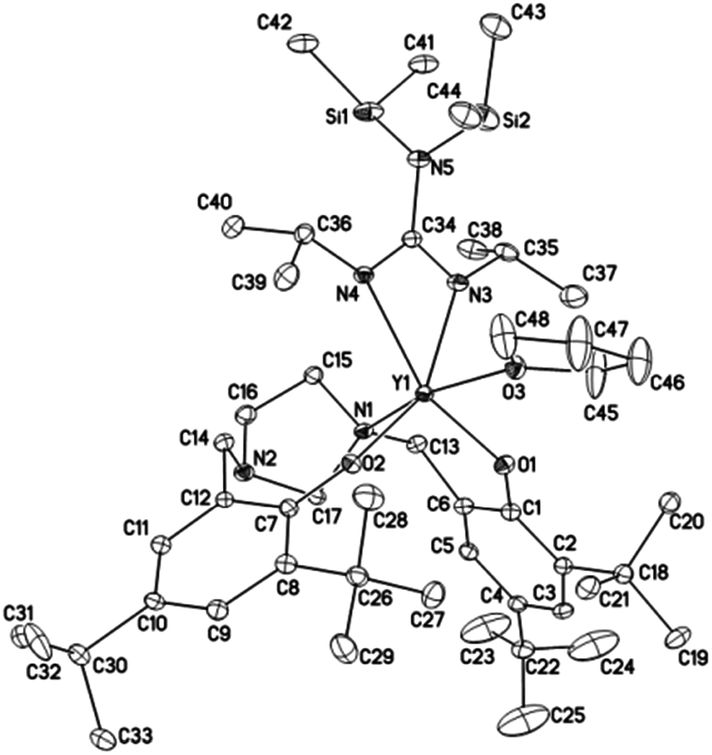 | ||
| Fig. 3 ORTEP diagram of complex 4 showing the atom numbering scheme. Thermal ellipsoids are drawn at the 10% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
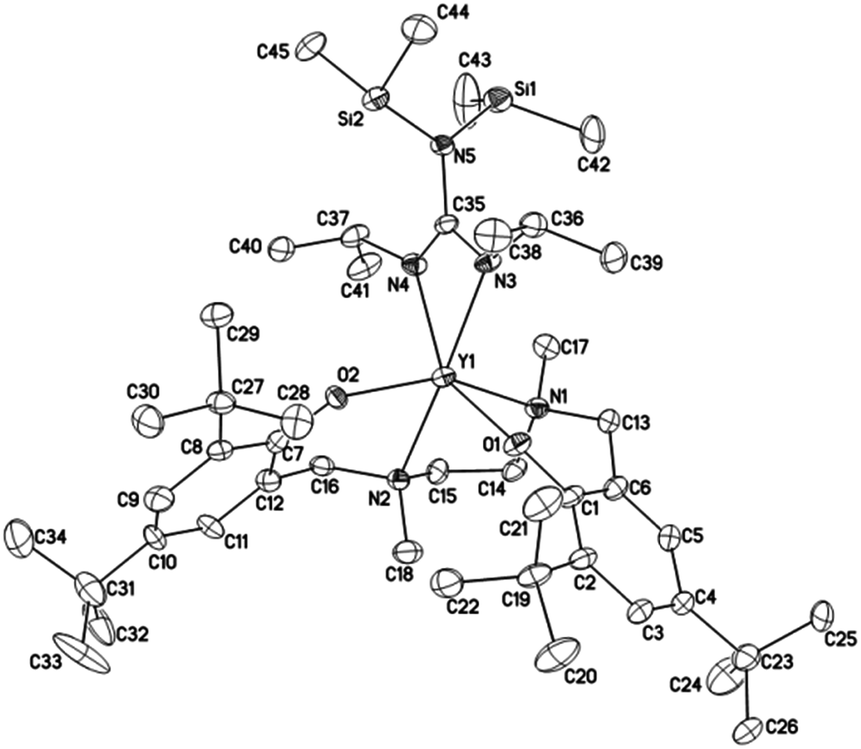 | ||
| Fig. 4 ORTEP diagram of complex 5 showing the atom numbering scheme. Thermal ellipsoids are drawn at the 10% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. | ||
| Compound | (2 × 1)·toluene | 2 | 4·toluene | (2 × 5)·THF |
|---|---|---|---|---|
| Formula | C101H172N10O4Yb2 | C46H78N5O2Yb | C55H94N5O3Si2Y | C94H172N10O5Si4Y2 |
| Formula weight | 1936.57 | 906.17 | 1018.44 | 1812.60 |
| T (K) | 223(2) | 223(2) | 223(2) | 223(2) |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Crystal size (mm) | 0.80 × 0.40 × 0.30 | 0.40 × 0.40 × 0.20 | 0.70 × 0.60 × 0.40 | 1.40 × 1.00 × 0.20 |
| Space group | P21/c | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/c |
| a (Å) | 10.8249(7) | 11.2280(7) | 12.0592(16) | 20.91(5) |
| b (Å) | 15.1480(10) | 11.8377(7) | 16.234(2) | 9.786(18) |
| c (Å) | 31.913(2) | 20.1349(18) | 30.021(4) | 28.19(9) |
| α (o) | 90 | 86.230(7) | 90 | 90 |
| β (o) | 98.688(2) | 82.991(7) | 90.787(4) | 102.70(7) |
| γ (o) | 90 | 64.118(4) | 90 | 90 |
| V (Å3) | 5172.9(6) | 2389.5(3) | 5876.6(13) | 5627(25) |
| Z | 2 | 2 | 4 | 2 |
| Dcalc. (g cm−3) | 1.243 | 1.259 | 1.151 | 1.070 |
| μ (mm−1) | 1.848 | 1.995 | 1.076 | 1.115 |
| F (000) | 2040 | 950 | 2200 | 1960 |
| θmax (o) | 27.49 | 27.49 | 27.48 | 27.49 |
| Collected | 29123 |
22816 |
32030 |
26518 |
| Unique reflns | 11753 |
10772 |
13342 |
12769 |
| Obsd reflns [I > 2.0σ(I)] | 10165 |
9609 | 8780 | 4141 |
| No. of variables | 532 | 500 | 581 | 546 |
| Goodness-of-fit | 1.132 | 1.087 | 1.099 | 1.045 |
| R | 0.0575 | 0.0529 | 0.1220 | 0.1471 |
| wR | 0.1242 | 0.0992 | 0.3109 | 0.3242 |
| Bond lengths | 1 | 2 | Bond lengths | 4 | Bond lengths | 5 |
|---|---|---|---|---|---|---|
| Yb1–O1 | 2.116(3) | 2.092(3) | Y1–O1 | 2.157(5) | Y1–O1 | 2.117(8) |
| Yb1–O2 | 2.132(3) | 2.105(3) | Y1–O2 | 2.141(4) | Y1–O2 | 2.093(9) |
| Yb1–N1 | 2.495(4) | 2.513(3) | Y1–O3 | 2.397(5) | Y1–N1 | 2.538(10) |
| Yb1–N2 | 2.554(4) | 2.553(4) | Y1–N1 | 2.514(5) | Y1–N2 | 2.574(9) |
| Yb1–N3 | 2.293(4) | 2.282(4) | Y1–N3 | 2.396(6) | Y1–N3 | 2.312(9) |
| Yb1–N4 | 2.321(4) | 2.338(4) | Y1–N4 | 2.406(5) | Y1–N4 | 2.418(10) |
| Bond angles | 1 | 2 | Bond angles | 4 | Bond angles | 5 |
|---|---|---|---|---|---|---|
| N2–Yb1–N3 | 165.4(15) | 163.3(12) | O1–Y1–O2 | 107.0(18) | O1–Y1–O2 | 105.4(3) |
| O1–Yb1–N1 | 78.6(14) | 78.8(11) | O1–Y1–O3 | 88.7(18) | O1–Y1–N1 | 79.2(3) |
| O1–Yb1–N4 | 101.9(15) | 100.3(13) | O2–Y1–O3 | 95.0(19) | O1–Y1–N2 | 95.3(3) |
| O2–Yb1–N1 | 78.9(13) | 78.1(11) | O1–Y1–N1 | 77.6(18) | O1–Y1–N3 | 96.7(3) |
| O2–Yb1–N4 | 100.7(15) | 103.1(13) | O1–Y1–N4 | 154.3(19) | O1–Y1–N4 | 150.7(3) |
| O1–Yb1–O2 | 157.0(14) | 155.8(11) | O2–Y1–N1 | 92.5(18) | O2–Y1–N1 | 147.4(3) |
| N1–Yb1–N4 | 179.2(15) | 177.3(12) | O2–Y1–N4 | 98.7(18) | N1–Y1–N2 | 70.8(3) |
| O1–Yb1–N2 | 90.4(15) | 88.2(13) | O3–Y1–N1 | 165.9(18) | N1–Y1–N3 | 100.3(4) |
| N3–Yb1–N4 | 58.0(15) | 58.2(13) | N1–Y1–N3 | 88.2(19) | N1–Y1–N4 | 93.6(3) |
| N3–Y1–N4 | 55.6(19) | N3–Y1–N4 | 56.2(3) |
In complexes 1 and 2, the coordinating environments around the ytterbium ions are similar. Same as other rare-earth metal complexes stabilized by the same ligand L1,10,17,22,24,38 the amino side-arm was also found to bind to the metal center in the solid state. The ytterbium centre is six-coordinated by two oxygen atoms and two nitrogen atoms from one amine-bridged bis(phenolate) ligand (L1), and two nitrogen atoms from one guanidinate group. The coordination geometry can be described as distorted octahedron, in which O1, O2, N1, and N4 atoms occupy equatorial positions, and N2 and N3 atoms occupy axial positions. The N2–Yb–N3 angles are found to be 163 and 165°, respectively.
The yttrium ion in complex 4 is six-coordinated by two oxygen atoms and one nitrogen atom from the imidazolidine-bridged bis(phenolate) ligand (L2), two nitrogen atoms from the guanidinate group and one oxygen atom from one THF molecule. It is noteworthy that only one nitrogen atoms from the imidazolidine ring binds to the metal center in the solid state, which is different from the yttrium amide bearing the same ligand L2.32
In complex 5, the yttrium center is hexacoordinated by two oxygen atoms and two nitrogen atoms from the bis(phenolate) ligand L3, and two nitrogen atoms from the guanidinate group.
The average Y–O(Ar) bond lengths in 5 is 2.105 Å, which is slightly shorter than those in 4 (2.149 Å) and L1-ligated yttrium guanidinates (2.128–2.162 Å).38 The bond angles of O1–Y–O2 in 4 (107.0(18)°) and 5 (105.4(3)°) are much smaller than the average angle of 153.2° in L1-ligated yttrium guanidinates. An obvious difference is also found for the values of the dihedral angle between the phenyl rings of the bis(phenolate) ligands, which are 84.5° for 4, and 81.8° for 5. Significantly larger values of 142.6–161.3° were found for L1-ligated yttrium guanidinates, which suggests that the N-heterocycle bridge in 4 and the ethylenediamine bridge in 5 result in a more distorted geometry of the complexes. Such a difference might contribute to the different activity and selectivity of these complexes in initiating ROP.
Polymerization of rac-LA initiated by complexes 1–8
In general, all complexes 1–8 have been found to be active initiators for the controlled ROP of rac-LA at room temperature in THF. More than 70% yields were obtained within 1 h with monomer-to-initiator ratios of up to 1000 at 25 °C. The results are summarized in Table 3.| Entry | Initiator | [M]0/[I]0 | Yieldb (%) | Mcc ( × 104) | Mnd ( × 104) | Đd | Pre |
|---|---|---|---|---|---|---|---|
| a General polymerization conditions: THF as the solvent, [rac-LA] = 1 mol L−1, at 25 °C, reaction time 1 h.b Yield: weight of polymer obtained/weight of monomer used.c Mc = (144.13) × [M]0/[I]0 × (polymer yield) (%).d Measured by GPC calibrated with standard polystyrene samples.e Pr is the probability of racemic linkages between monomer units and is determined from the methine region of the homonuclear decoupled 1H NMR spectrum.f In CH2Cl2.g In toluene. | |||||||
| 1 | 1 | 500 | 99 | 7.1 | 8.9 | 1.16 | 0.99 |
| 2f | 1 | 500 | 67 | 4.8 | 6.9 | 1.25 | 0.78 |
| 3g | 1 | 500 | 99 | 7.1 | 8.6 | 1.23 | 0.75 |
| 4 | 1 | 300 | 99 | 4.3 | 5.2 | 1.15 | 0.98 |
| 5 | 1 | 700 | 99 | 10.0 | 11.5 | 1.12 | 0.98 |
| 6 | 1 | 1000 | 98 | 14.1 | 15.1 | 1.13 | 0.99 |
| 7 | 1 | 1200 | 98 | 16.9 | 17.8 | 1.11 | 0.98 |
| 8 | 1 | 1500 | 84 | 18.2 | 18.8 | 1.15 | 0.97 |
| 9 | 2 | 1000 | 97 | 14.0 | 13.2 | 1.08 | 0.99 |
| 10 | 2 | 1500 | 75 | 16.2 | 15.1 | 1.08 | 0.98 |
| 11 | 3 | 500 | 97 | 7.0 | 8.8 | 1.23 | 0.98 |
| 12 | 3 | 1000 | 90 | 13.0 | 16.5 | 1.21 | 0.97 |
| 13 | 4 | 500 | 83 | 6.0 | 8.5 | 1.68 | 0.62 |
| 14 | 4 | 1000 | 80 | 11.5 | 14.6 | 1.65 | 0.62 |
| 15 | 5 | 500 | 76 | 5.5 | 3.4 | 1.36 | 0.69 |
| 16 | 5 | 1000 | 70 | 10.1 | 12.0 | 1.35 | 0.69 |
| 17 | 6 | 500 | 95 | 6.9 | 4.2 | 1.44 | 0.51 |
| 18 | 6 | 1000 | 92 | 13.3 | 14.8 | 1.41 | 0.52 |
| 19 | 7 | 500 | 95 | 6.8 | 3.1 | 1.66 | 0.82 |
| 20 | 7 | 1000 | 91 | 13.1 | 15.3 | 1.57 | 0.82 |
| 21 | 8 | 500 | 94 | 6.7 | 3.3 | 1.58 | 0.82 |
| 22 | 8 | 1000 | 90 | 13.0 | 15.9 | 1.63 | 0.82 |
Using complex 1 as the initiator, the ROP of rac-LA proceeds faster in THF and toluene than in CH2Cl2, and with higher stereoselectivities in THF than in toluene and CH2Cl2 (Table 3, entries 1–3). Overall, THF is the optimal solvent, which is consistent with reported results.24 Almost quantitative yields were obtained with the ratio of monomer to complex 1 increasing from 300 to 1200 (Table 3, entries 4–7). A good yield of 84% was still obtained when the ratio was increased to 1500. Moreover, highly heterotactic PLA (Pr ≥ 0.97) was obtained. Complexes 1, 2 and 3 bearing the same bis(phenolate) ligand L1 showed similar activities (Table 3, entries 6, 9 and 12), while complex 1 worked slightly better than the other two in the case of high [M]0/[I]0 ratios (Table 3, entries 7, 8, 10, and 12). Moreover, Pr values of the polymers are essentially the same. These findings are similar to results from lanthanide alkyls, aryloxides and oxides stabilized by the same phenolate ligand.17–19,22,24 Thus, neither the metal center nor the guanidinate group plays significant roles in influencing the polymerization rates or tacticity.
A comparative study of complexes 4–8 on the activity in initiating the stereoselective ROP of rac-LA under identical conditions revealed substantial differences (Table 3). Using complex 4 as the initiator, the yield is 83% and Pr is 0.62 when the ratio of monomer to initiator is 500 (Table 3, entry 13), which is similar to that of yttrium amide bearing the same imidazolidine-bridged bis(phenolate) ligand.21,32 Complex 5 initiated ROP of rac-LA and afforded PLA with Pr value of 0.69 (Table 3, entries 15 and 16). The alkyl analogue of 5 (ref. 17) was reported to initiate ROP of rac-LA and afforded PLA of almost the same Pr value. Complex 6 effectively initiated rac-LA polymerization and produced PLA with Pr value of 0.51 (Table 3, entries 17 and 18). The stereoselectivity of 6 is similar to that of homopiperazine-bridged bis(phenolate) Ti(IV) complex.42 The methoxy-amine-bis(phenolate) yttrium complex 7 showed the same level of activity and selectivity (Pr = 0.82) as furan-amine-bis(phenolate) yttrium complexes 8 (Table 3, entries 19–22). Based on the findings discussed above, it is conclusive that the bis(phenolate) ligands exert crucial roles in influencing the tacticity. Overall, complexes stabilized by ligands with coordinating arms (1, 2, 3, 7, and 8) show higher stereoselectivity (Pr values of 0.82–0.99), while those carrying ligands of longer bridges (4, 5, and 6) show lower stereoselectivity (Pr values of 0.51–0.69). This finding emphasizes again the importance of bis(phenolate) ligands on tacticity.8f,8u,9,23–26
All PLA obtained in the presence of complexes 1–8 showed relatively narrow molecular weight distributions in the range of Mw/Mn = 1.08–1.68. It is noteworthy that the average-number molecular weights (Mn) values of PLA obtained with complexes 1–3 are generally in good agreement with theoretical Mn values, which indicate that the polymerization proceeded in a living fashion without other significant side reactions. To illustrate this living character, the relationship between the number-average molecular weight (Mn) and the molar ratio of monomer to initiator 1 ([M]0/[I]0) was plotted in Fig. 5. As the monomer/initiator ratio increases, the molecular weight of the resulting polymer increased linearly, while molecular weight distributions were kept almost unchanged (1.12–1.16), strongly suggesting that the polymerization process is controllable.
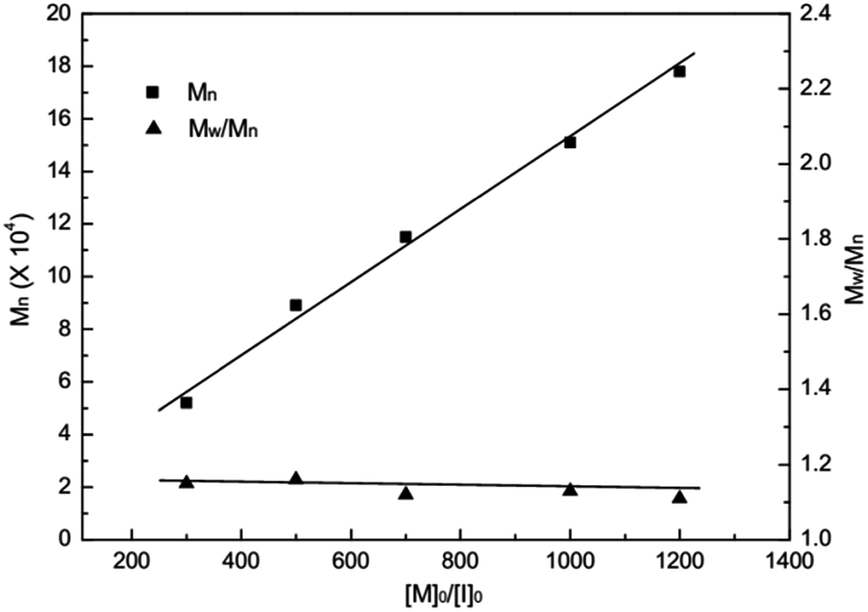 | ||
| Fig. 5 Polymerization of rac-LA initiated by complex 1 in THF at 25 °C. Relationship between the number-averaged molecular weight (Mn) and the molar ratio of monomer to initiator. | ||
To investigate the mechanism of ROP of rac-LA initiated by these guanidinate complexes, oligomerization of rac-LA initiated by complex 1 in a [M]0/[I]0 ratio of 15 was carried out. However, no guanidinate or bis(phenolate) group was identified from the 1H NMR spectrum of the oligomer. Considering the instability of the oligomer with the guanidinate end cap,38 a new oligomer was prepared by quenching the oligomerization described above with benzyl alcohol. End group analysis by 1H NMR spectroscopy clearly shows the existence of a benzyloxy group and a HOCH(CH3)CO– group according to the resonances at about 5.18, 7.33 ppm assignable to the former and at 1.23, 2.57 and 4.34 ppm assignable to the latter, as shown in Fig. 6. The benzyloxy group is believed to come from the exchange reaction of benzyl alcohol with the guanidinate group. Meanwhile, no resonances corresponding to the bis(phenolate) ligand L1 was observed in the 1H NMR spectrum, which ruled out the possibility that the amine-bridged bis(phenolate) group is the initiating group in the polymerization process. These results revealed that the guanidinate group in these complexes acted as the initiating group in the ROP of rac-LA, while the bis(phenolate) group works as the spectator ligand controlling the stereoselectivity of the polymerization process.24,25
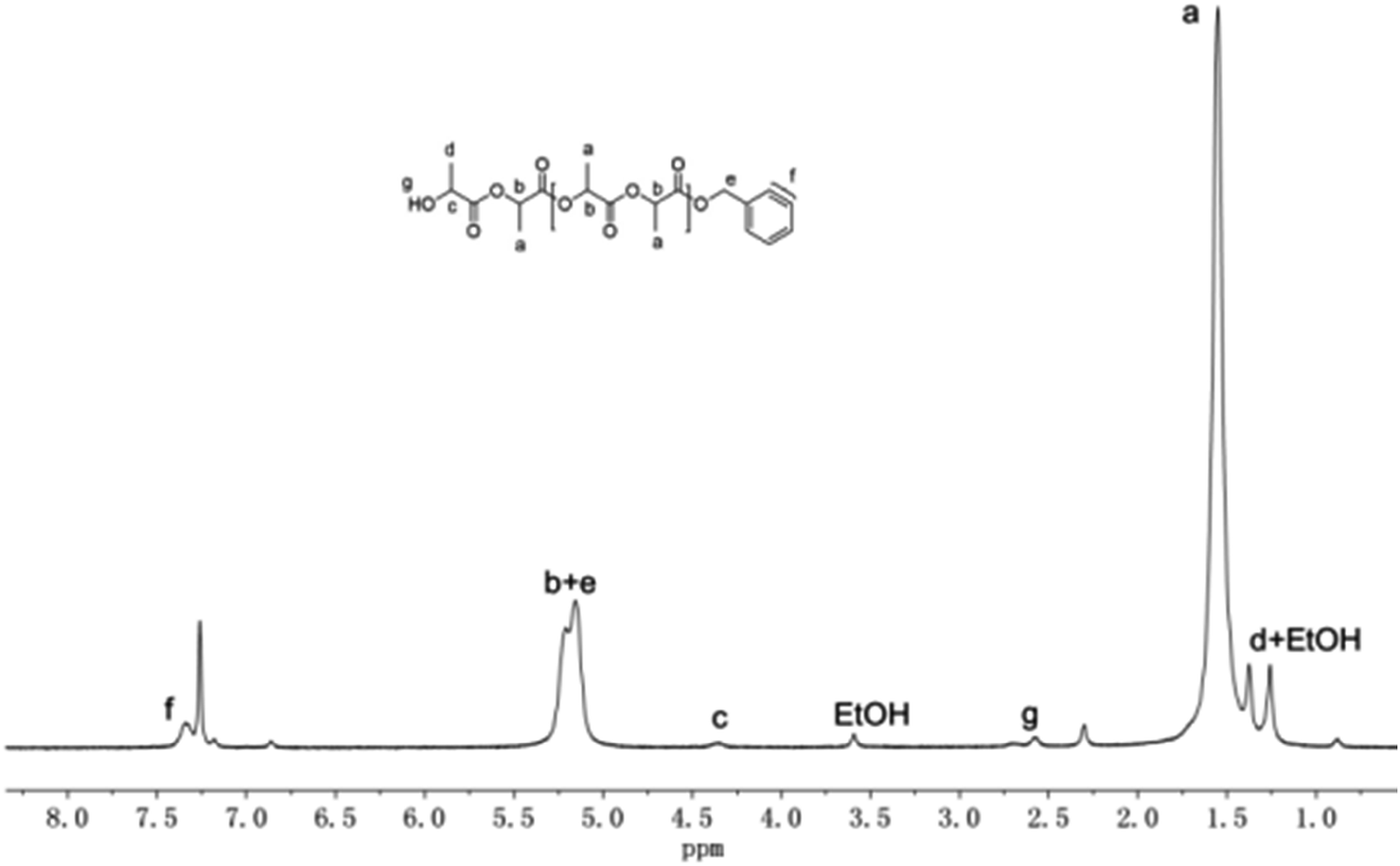 | ||
| Fig. 6 1H NMR spectrum of rac-LA oligomer initiated by complex 1 after quenching with benzyl alcohol. | ||
ROP of rac-BBL initiated by complexes 1–8
ROP of BBL is generally much more challenging than that of LA despite of their larger ring strains,43 and most of the catalysts show low stereoselectivity and a high prevalence of chain termination events, such as transesterification and chain transfer.41,44 Complexes 1–8 proved efficient in initiating the ROP of rac-BBL, and are in general capable of controlling stereoselectivity to produce highly syndiotactic PHB in toluene. Representative results are summarized in Table 4.| Entry | Initiator | [M]0/[I]0 | t | Yieldb (%) | Mcc (× 104) | Mnd (× 104) | Đd | Pre |
|---|---|---|---|---|---|---|---|---|
| a General polymerization conditions: toluene as the solvent, [rac-BBL] = 2 mol L−1, at 25 °C.b Yield: weight of polymer obtained/weight of monomer used.c Mc = 86.09 × [M]0/[I]0 × (polymer yield) (%).d Measured by GPC calibrated with standard polystyrene samples.e Pr is the probability of racemic linkages between monomer units and is determined from the carbonyl region of the 13C{1H} NMR spectroscopy at 25 °C.f In THF.g [rac-BBL] = 1 mol L−1. | ||||||||
| 1 | 1 | 200 | 2 min | 100 | 1.7 | 2.5 | 1.35 | 0.82 |
| 2 | 1 | 400 | 10 min | 98 | 3.4 | 3.5 | 1.33 | 0.82 |
| 3 | 1 | 600 | 10 min | 98 | 5.1 | 6.5 | 1.31 | 0.82 |
| 4 | 1 | 800 | 10 min | 98 | 6.7 | 8.2 | 1.36 | 0.82 |
| 5 | 1 | 1000 | 10 min | 96 | 8.3 | 9.7 | 1.42 | 0.82 |
| 6 | 1 | 1500 | 30 min | 88 | 11.4 | 13.5 | 1.37 | 0.82 |
| 7f | 1 | 400 | 30 min | 60 | 2.1 | 3.0 | 1.35 | 0.67 |
| 8g | 1 | 400 | 10 min | 83 | 2.9 | 3.3 | 1.30 | 0.82 |
| 9 | 2 | 400 | 10 min | 100 | 3.4 | 4.2 | 1.29 | 0.82 |
| 10 | 2 | 1000 | 10 min | 92 | 7.9 | 9.7 | 1.44 | 0.82 |
| 11 | 3 | 200 | 1 h | 97 | 1.7 | 2.5 | 1.35 | 0.82 |
| 12 | 3 | 400 | 1 h | 80 | 2.4 | 3.1 | 1.31 | 0.82 |
| 13 | 4 | 200 | 10 h | 70 | 1.2 | 2.3 | 1.78 | 0.58 |
| 14 | 4 | 400 | 10 h | 55 | 1.9 | 3.0 | 1.86 | 0.58 |
| 15 | 5 | 200 | 10 h | 16 | 0.3 | — | — | — |
| 16 | 6 | 200 | 10 h | 96 | 1.7 | 1.8 | 1.83 | 0.57 |
| 17 | 6 | 400 | 10 h | 85 | 2.9 | 3.5 | 1.78 | 0.57 |
| 18 | 7 | 200 | 10 h | 96 | 1.7 | 1.7 | 1.86 | 0.81 |
| 19 | 7 | 400 | 10 h | 75 | 2.6 | 3.3 | 1.80 | 0.81 |
| 20 | 8 | 200 | 10 h | 63 | 1.1 | 2.3 | 1.78 | 0.79 |
| 21 | 8 | 400 | 10 h | 56 | 1.9 | 3.2 | 1.82 | 0.79 |
A full conversion of rac-BBL was achieved within 2 min in the ratio of [M]0/[I]0 = 200 (Table 4, entry 1) in toluene at 25 °C. Moreover, highly syndiotactic PHB (Pr = 0.82) was obtained. Increasing the monomer to initiator ratio to 1000 still led to almost full conversion within 10 min (Table 4, entries 2–5). A lower of yield of 88% was obtained when the monomer to initiator ratio was raised to 1500 (Table 4, entry 6). The solvent plays a significant role in influencing the activity and stereoselectivity, as the ROP proceeded much slower in THF and afforded PHB of lower Pr value (Table 4, entries 2 and 7). Decreasing the monomer concentration also slowed down the polymerization rate (Table 4, entries 2 and 8). These findings are consistent with those observed in the amino-bridged bis(phenolate) lanthanide systems.14,15,24
Complex 2 showed similar activities and stereoselective control in initiating the ROP of rac-BBL (Table 4, entries 9 and 10). Both ytterbium complexes 1 and 2 are more active than the yttrium complex 3 (Table 4, entries 11 and 12), which may be attributed to the rare-earth metal centres of different ionic radii. However, the stereoselectivity is not at all influenced, which is quite different from that observed in rac-LA polymerization (vide supra).
The activities and stereoselective control of complexes 4–8 in initiating the ROP of rac-BBL were examined and compared under identical conditions. Complex 4 bearing an imidazolidine-bridged bis(phenolate) ligand is less active than complex 3, and the Pr value of the resulting PHB is much lower (Table 4, entries 13 and 14). The activity of complex 5 (Table 4, entry 15) is the lowest among complexes 1–8, and is obviously lower than that of the analogous yttrium amide.9 Complex 6 bearing a homopiperazine-bridged bis(phenolate) ligand showed higher activity than that of 4, while resulting PHB have almost the same Pr values (Table 4, entries 16 and 17). The methoxy-amine-bis(phenolate) complex 7 was found to be more active than the furan-amine-bis(phenolate) complex 8, and these two complexes show similar stereoselective control (Pr = 0.79–0.81). The same influence exerted by bis(phenolate) ligands on stereoselectivity has been observed as that for polymerization of rac-lactide (vide supra).
It was found that the PHB obtained with complexes 1–3 had relatively narrow molecular weight distributions (1.13 < Mw/Mn < 1.44) and average-number molecular weights (Mn) are generally in good agreement with the theoretical Mn values. For the ROP of rac-BBL initiated by complex 1, the number-average molecular masses (Mn) values of the resultant PHB increases as the monomer-to-initiator ratio increases, while the values of Mw/Mn were kept almost unchanged, indicating that the polymerization process is also controllable (Table 4, entries 1–5). However, the PHB produced with 4–8 in toluene had relatively broad molecular weight distributions (Mw/Mn = 1.31–1.86), which may be mainly attributed to the transesterification reactions during the polymerization process or to the relatively slow initiation step.
The ROP mechanism of rac-BBL was elucidated by using the method similar to that of rac-LA. The oligomer of rac-BBL was prepared by the reaction of complex 1 with rac-BBL in a 1:20 molar ratio which is quenched by benzyl alcohol. In addition to the resonances characteristic for the HOCH(CH3)CH2CO– group at 4.19, 2.30 and 1.38 ppm, respectively, the 1H NMR spectrum in CDCl3 clearly showed the existence of the benzyloxy group according to the resonances at about 7.34 and 5.12 ppm (Fig. 7), which arises from the exchange reaction between benzyl alcohol with the guanidinate group. Meanwhile, additional resonances at 6.95 and 5.81 ppm were observed, which may correspond to trans-crotonate groups that may result from the elimination side reactions in the polymerization progress. A similar elimination reaction in rac-BBL polymerization has been observed using amino-bridged bis(phenolate)lanthanide alkoxo complexes as the initiators.24
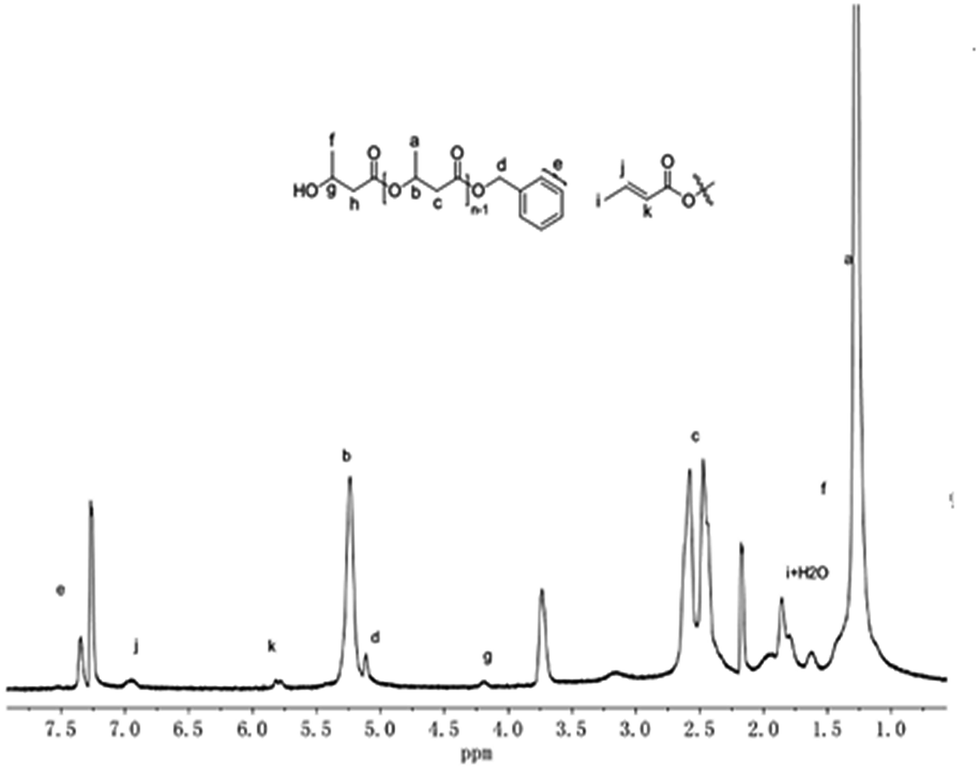 | ||
| Fig. 7 1H NMR spectrum of rac-BBL oligomer initiated by complex 1 after quenching with benzyl alcohol. | ||
Conclusion
In summary, eight rare-earth metal guanidinates supported by a series of amine-bridged bis(phenolate) ligands were synthesized according to either metathesis reactions, or insertion reactions of carbodiimides. All eight complexes have been fully characterized, and the solid state structures of four of them have been determined. A comparative study on these complexes in initiating the ROP of rac-LA and rac-BBL revealed that bis(phenolate) ligands play critical roles in governing the stereoselectivity. ROP initiated by complexes 1–3 bearing the amine-bridged bis(phenolate) ligand L1 afforded highly heterotactic PLA (Pr values of more than 0.97) and syndiotactic PHB (Pr values of 0.82). End group analysis of the oligomer revealed that in the ROP of rac-LA and rac-BBL the guanidinate groups serve as initiating groups.Experimental
Reagents and general procedures
All manipulations requiring dry atmosphere were performed under a purified argon atmosphere by use of standard Schlenk techniques or in a glovebox. Solvents were degassed and distilled from sodium benzophenone ketyl under argon prior to use. Ligands L1H2–L6H2,38 lanthanide precursors L1YbCl(THF)30,45 and L1Y[N(SiHMe2)2](THF) to L6Y[N(SiHMe2)2](THF)9 were prepared by modifications of the methods reported in the literature. Benzyl alcohol were dried over 4 Å molecular sieves for 1 week and then distilled before use. rac-Lactide (rac-LA) was recrystallized twice from dry toluene and then sublimed under vacuum at 50 °C. rac-β-Butyrolactone (rac-BBL) was freshly distilled from CaH2 under nitrogen and degassed thoroughly by freeze–pump–thaw cycles prior to use.Lanthanide analyses were performed by ethylenediaminetetraacetic acid titration with a xylenol orange indicator and a hexamine buffer. Carbon, hydrogen, and nitrogen analyses were performed by direct combustion with a Carlo-Erba EA-1110 instrument. The IR spectra were recorded with a Nicolet-550 Fourier transform IR spectrometer as KBr pellets. The 1H and 13C NMR spectra were recorded in a C6D6 solution for complexes 3–6 with a Unity Varian spectrometer. Molecular weights and molecular weight distributions were determined against polystyrene standards by gel permeation chromatography (GPC) on a PL 50 apparatus, and THF was used as an eluent at a flow rate of 1.0 mL min−1 at 40 °C. The microstructures of PLAs and PHBs were measured by homodecoupling 1H NMR spectroscopy at 20 °C in CDCl3 and by 13C{1H} NMR spectroscopy at 40 °C in CDCl3, respectively, on a Unity Varian AC-400 spectrometer.
General polymerization procedure
The procedures for polymerization of rac-LA and rac-BBL were similar and given below:A 20 mL vial, equipped with a magnetic stirring bar, was charged in a glovebox with the desired amount of monomer and solvent. After the monomer was dissolved, a solution of the initiator was added to this solution via a syringe. The mixture was immediately stirred at the desired temperature for the desired time. The reaction was quenched by the addition of ethanol and then poured into ethanol to precipitate the polymer, which was dried under vacuum to constant weight.
Oligomer preparation
Oligomerization of rac-LA and rac-BBL was carried out with complex 1 as the initiator in THF and toluene, respectively, at 25 °C under the condition of a molar ratio of [monomer]/[initiator] of 15 or 20. The polymerization mixture was stirred for 0.5 h and then quenched by adding benzyl alcohol. The precipitated oligomers were collected, dried under vacuum, and used for 1H NMR measurement.X-ray crystallographic structure determinations
Suitable single crystals of complexes 1, 2, 4 and 5 were sealed in a thin-walled glass capillary for determination of the single-crystal structures. Intensity data were collected with a Rigaku Mercury CCD area detector in ω scan mode using Mo Kα radiation (λ = 0.71070 Å). The diffracted intensities were corrected for Lorentz/polarization effects and empirical absorption corrections.The structures were solved by direct methods and refined by full-matrix least-squares procedures based on |F|2. The hydrogen atoms in these complexes were generated geometrically, assigned appropriate isotropic thermal parameters, and allowed to ride on their parent carbon atoms. All of the hydrogen atoms were held stationary and included in the structure factor calculation in the final stage of full matrix least-squares refinement. The structures were solved and refined using SHELXTL-97 programs.
Acknowledgements
Financial support from the National Natural Science Foundation of China (Grants 21174095, 21132002, 21172165, 21372172 and 21402135), PAPD, the Major Research Project of the Natural Science Foundation of the Jiangsu Higher Education Institutions (Project 14KJA150007),and the Qing Lan Project is gratefully acknowledged.Notes and references
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- S. Mecking, Angew. Chem., Int. Ed., 2004, 43, 1078–1085 CrossRef CAS PubMed.
- A. P. Dove, Chem. Commun., 2008, 48, 6446–6470 RSC.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS PubMed.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed.
- J. Yang, R. van Lith, K. Baler, R. A. Hoshi and G. A. Ameer, Biomacromolecules, 2014, 15, 3942–3952 CrossRef CAS PubMed.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- (a) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed; (b) A. Amgoune, C. M. Thomas and J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS PubMed; (c) N. Ajellal, D. M. Lyubov, M. A. Sinenkov, G. K. Fukin, A. V. Cherkasov, C. M. Thomas, J.-F. Carpentier and A. A. Trifonov, Chem.–Eur. J., 2008, 14, 5440–5448 CrossRef CAS PubMed; (d) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2010, 29, 491–500 CrossRef CAS; (e) E. Grunova, E. Kirillov, T. Roisnel and J.-F. Carpentier, Dalton Trans., 2010, 6739–6752 RSC; (f) M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem.–Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed; (g) Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed; (h) H. Y. Ma and J. Okuda, Macromolecules, 2005, 38, 2665–2673 CrossRef CAS; (i) H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef PubMed; (j) H. Ma, T. P. Spaniol and J. Okuda, Inorg. Chem., 2008, 47, 3328–3339 CrossRef CAS PubMed; (k) B. Lian, H. Ma, T. P. Spaniol and J. Okuda, Dalton Trans., 2009, 9033–9042 RSC; (l) J. C. Buffet, A. Kapelski and J. Okuda, Macromolecules, 2010, 43, 10201–10203 CrossRef CAS; (m) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS; (n) H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32–35 RSC; (o) L. Clark, M. G. Cushion, H. E. Dyer, A. D. Schwarz, R. Duchateau and P. Mountford, Chem. Commun., 2010, 46, 273–275 RSC; (p) H. E. Dyer, S. Huijser, N. Susperregui, F. Bonnet, A. D. Schwarz, R. Duchateau, L. Maron and P. Mountford, Organometallics, 2010, 29, 3602–3621 CrossRef CAS; (q) T.-P.-A. Cao, A. Buchard, X. F. Le Goff, A. Auffrant and C. K. Williams, Inorg. Chem., 2012, 51, 2157–2169 CrossRef CAS PubMed; (r) C. Bakewell, R. H. Platel, S. K. Cary, S. M. Hubbard, J. M. Roaf, A. C. Levine, A. J. P. White, N. J. Long, M. Haaf and C. K. Williams, Organometallics, 2012, 31, 4729–4736 CrossRef CAS; (s) E. M. Broderick, P. S. Thuy-Boun, N. Guo, C. S. Vogel, J. Sutter, J. T. Miller, K. Meyer and P. L. Diaconescu, Inorg. Chem., 2011, 50, 2870–2877 CrossRef CAS PubMed; (t) E. M. Broderick, N. Guo, C. S. Vogel, C. L. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed; (u) A. Amgoune, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Chem.–Eur. J., 2006, 12, 169–179 CrossRef CAS PubMed.
- J. W. Kramer, D. S. Treitler, E. W. Dunn, P. M. Castro, T. Roisnel, C. M. Thomas and G. W. Coates, J. Am. Chem. Soc., 2009, 131, 16042–16044 CrossRef CAS PubMed.
- C. X. Cai, L. Toupet, C. W. Lehmann and J.-F. Carpentier, J. Organomet. Chem., 2003, 683, 131–136 CrossRef CAS.
- Y. Chapurina, J. Klitzke, O. D. L. Casagrande Jr, M. Awada, V. Dorcet, E. Kirillov and J.-F. Carpentier, Dalton Trans., 2014, 14322–14333 RSC.
- J. S. Klitzke, T. Roisnel, E. Kirillov, O. D. L. Casagrande Jr and J.-F. Carpentier, Organometallics, 2014, 33, 309–321 CrossRef CAS.
- C. X. Cai, A. Amgoune, C. W. Lehmann and J.-F. Carpentier, Chem. Commun., 2004, 330–331 RSC.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed.
- N. Ajellal, M. Bouyahyi, A. Amgoune, C. M. Thomas, A. Bondon, I. Pillin, Y. Grohens and J.-F. Carpentier, Macromolecules, 2009, 42, 987–993 CrossRef CAS.
- F. Bonnet, A. R. Cowley and P. Mountford, Inorg. Chem., 2005, 44, 9046–9055 CrossRef CAS PubMed.
- X. Liu, X. Shang, T. Tang, N. Hu, F. Pei, D. Cui, X. Chen and X. Jing, Organometallics, 2007, 26, 2747–2757 CrossRef CAS.
- W. Zhao, C. Li, B. Liu, X. Wang, P. Li, Y. Wang, C. Wu, C. Yao, T. Tang, X. Liu and D. Cui, Macromolecules, 2014, 47, 5586–5594 CrossRef CAS.
- W. Zhao, D. Cui, X. Liu and X. Chen, Macromolecules, 2010, 43, 6678–6684 CrossRef CAS.
- W. Zhao, Y. Wang, X. Liu and D. Cui, Chem. Commun., 2012, 48, 4483–4485 RSC.
- Y. Luo, W. Li, D. Lin, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2010, 29, 3507–3514 CrossRef CAS.
- K. Nie, X. Gu, Y. Yao, Y. Zhang and Q. Shen, Dalton Trans., 2010, 6832–6840 RSC.
- W. Li, Z. Zhang, Y. Yao, Y. Zhang and Q. Shen, Organometallics, 2012, 31, 3499–3511 CrossRef CAS.
- K. Nie, L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Inorg. Chem., 2012, 51, 11133–11143 CrossRef CAS PubMed.
- K. Nie, T. Feng, F. Song, Y. Zhang, H. Sun, D. Yuan, Y. Yao and Q. Shen, Sci. China: Chem., 2014, 57, 1106–1116 CrossRef.
- (a) S. Yang, K. Nie, Y. Zhang, M. Xue, Y. Yao and Q. Shen, Inorg. Chem., 2013, 53, 105–115 CrossRef PubMed; (b) S. Yang, Z. Du, Y. Zhang and Q. Shen, Chem. Commun., 2012, 48, 9780–9782 RSC.
- M. Zhang, X. Ni and Z. Shen, Organometallics, 2014, 33, 6861–6867 CrossRef CAS.
- (a) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 9226–9230 CrossRef CAS PubMed; (b) M. Mazzeo, R. Tramontano, M. Lamberti, A. Pilone, S. Milione and C. Pellecchina, Dalton Trans., 2013, 9338–9351 RSC; (c) C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed; (d) Z. Mou, B. Liu, X. Liu, H. Xie, W. Rong, L. Li, S. Li and D. Cui, Macromolecules, 2014, 47, 2233–2241 CrossRef CAS; (e) M. Sinenkov, E. Kirillov, T. Roisnel, G. Fukin, A. Trifonov and J.-F. Carpentier, Organometallics, 2011, 30, 5509–5523 CrossRef CAS; (f) G. Li, M. Lamberti, M. Mazzeo, D. Pappalardo, G. Roviello and C. Pellecchia, Organometallics, 2012, 31, 1180–1188 CrossRef CAS; (g) C. Bakewell, T.-P.-A. Cao, X. F. Le Goff, N. J. Long, A. Auffrant and C. K. Williams, Organometallics, 2013, 32, 1475–1483 CrossRef CAS; (h) N. Maudoux, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5740–5748 CrossRef CAS.
- Y. Yao, M. Ma, X. Xu, Y. Zhang, Q. Shen and W.-T. Wong, Organometallics, 2005, 24, 4014–4020 CrossRef CAS.
- Z. Zhang, X. Xu, W. Li, Y. Yao, Y. Zhang, Q. Shen and Y. Luo, Inorg. Chem., 2009, 48, 5715–5724 CrossRef CAS PubMed.
- E. E. Delbridge, D. T. Dugah, C. R. Nelson, B. W. Skelton and A. H. White, Dalton Trans., 2007, 143–153 RSC.
- F. M. Kerton, A. C. Whitwood and C. E. Willans, Dalton Trans., 2004, 2237–2244 RSC.
- Z. Zhang, X. Xu, S. Sun, Y. Yao, Y. Zhang and Q. Shen, Chem. Commun., 2009, 7414–7416 RSC.
- Y. Yao, X. Xu, B. Liu, Y. Zhang, Q. Shen and W.-T. Wong, Inorg. Chem., 2005, 44, 5133–5140 CrossRef CAS PubMed.
- X. Xu, Y. Yao, M. Hu, Y. Zhang and Q. Shen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4409–4419 CrossRef CAS PubMed.
- L. Zhou, Y. Wang, Y. Yao, Y. Zhang and Q. Shen, J. Rare Earths, 2007, 25, 544–548 CrossRef.
- Q. Sun, Y. Wang, D. Yuan, Y. Yao and Q. Shen, Organometallics, 2014, 33, 994–1001 CrossRef CAS.
- T. Zeng, Y. Wang, Q. Shen, Y. Yao, Y. Luo and D. Cui, Organometallics, 2014, 33, 6803–6811 CrossRef CAS.
- T. Zeng, Q. Qian, Y. Wang, Y. Yao and Q. Shen, J. Organomet. Chem., 2015, 779, 14–20 CrossRef CAS PubMed.
- L. Zhou, Y. Yao, Y. Zhang, M. Xue, J. Chen and Q. Shen, Eur. J. Inorg. Chem., 2004, 2167–2172 CrossRef CAS PubMed.
- J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS PubMed.
- S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 2033–2037 RSC.
- A. Duda, A. Kowalski, P. Dubois, O. Coulembier and J. M. Raquez, Handb. Ring-Opening Polym., 2009, 1–51 CAS.
- C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC.
- C. E. Willans, M. A. Sinenkov, G. K. Fukin, K. Sheridan, J. M. Lynam, A. Trifonov and F. M. Kerton, Dalton Trans., 2008, 3592–3598 RSC.
Footnote |
| † CCDC 1047907–1047909 and 1048302 (for complex 1, 2, 4 and 5). For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra10151d |
| This journal is © The Royal Society of Chemistry 2015 |