
Novel organic–inorganic hybrids based on T8 and T10 silsesquioxanes: synthesis, cage-rearrangement and properties†
Mateusz Janeta,
Łukasz John*,
Jolanta Ejfler and
Sławomir Szafert
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie, 50-383 Wrocław, Poland. E-mail: lukasz.john@chem.uni.wroc.pl
First published on 18th August 2015
Abstract
In this paper, we present a simple approach for the synthesis of well-defined macromolecules based on precisely isolated amino- and amido-functionalized octa T8 and deca T10 silsesquioxanes (SQs). Here, we show that reorganization of the siloxane cage-like core (T8 → T10) can be easily performed, including isolation of intermediates, and cage rearrangement achieved by using Brønsted superacid, trifluoromethanesulfonic acid (CF3SO3H). Moreover, T10-like SQs can be obtained in a one-step reaction by alkoxysilane condensation in trifluoromethanesulfonic acid conditions. The resulting decamers of amine-SQ and an amido-functionalized derivative containing long alkyl chains are reported for the first time in the literature. The non-fluorinated amido derivatives due to their lamellar-like nature and specific packing can serve as transparent hydrophobic coatings in various industrial applications. The obtained compounds were fully characterized using FT-IR, UV-vis, multinuclear NMR (1H, 13C, 29Si), DOSY NMR, TG-DTA, DSC, HR-MS, TEM, EDS and elemental analysis.
Introduction
The development of new materials, including coatings and films, with specified structure, topology, transparency and variable hydrophobicity continues to be an intensively explored area of modern materials science, as mankind persistently tries to mimic the kaleidoscopic forms of nature. One such source of inspiration is natural hydrophobic surfaces.1 Hydrophobicity is a remarkable property that is strongly determined by the properties and structure of the so-called near-surface layer a few nanometers thick rather than by the characteristics of the bulky material. Such systems possess a set of functional properties, namely, they are water repellent, corrosion resistant and stable against biofouling and inorganic and, in some cases, organic pollutants; in the case of superhydrophobic systems they form self-cleaning coatings.2 Because of these unique features, such materials are of great interest and are widely used in our daily life.In the literature, there are numerous hydrophobic materials and coatings which cannot all be mentioned here. Most of them constitute multifunctional composites, blends, and other mixtures. They can be divided into two general groups: first, metal, mixed-metal and metal oxide-based materials; and second, expensive but efficient fluorinated organics and organometallics that constitute the largest group. For instance, Ganesh et al. fabricated hydrophobic coating using the electrospinning method on glass substrate with rice-shaped titanium dioxide nano/mesostructures.3 Xiong et al. used bifunctional silica particles and epoxy glue bearing poly(2-perfluorooctylethyl methacrylate) and poly(acrylic acid) coronal chains to fabricate hydrophobic coatings.4 Also, a mixture of bisphenol diglycidyl ether, tetraethyl orthosilicate and fluorinated side chain (F-silicon)-containing alkoxy silane deposited on silicon wafers forms hydrophobic epoxy coating.5 Choi et al. fabricated fluoro-based fibers, forming a web-like structure which was exhibited by an electrospun web of poly(2,2,2-trifluoroethyl methacrylate).6
It is worth noting that fabrication of the above-mentioned materials requires specific and not readily available techniques, complex technology and expensive fluorinated chemicals. It is also better, from the technological point of view, to use a well-defined compound as a hydrophobic agent rather than a multiple mixture. From this perspective, an attractive group is the cage-like silsesquioxanes (SQs) of the general formula (RSiO1.5)n (where n is an integer and R is an organic group). Covalently bonded organic side-chains make reactive functionalities suitable for polymerization or grafting and can be arbitrarily designed to achieve the desired affinity with the host silicon-based three-dimensional cage framework. SQ-based materials are commonly used as additives, plastics, carriers, preceramics, etc. Their use results in higher temperature stability and enhanced physical properties of the compositions. Polymers containing SQs show delayed combustion and improved mechanical properties. Because of the many benefits that can be derived from cage-like silsesquioxanes' properties, their chemistry has recently received a great deal of attention. From among fully functionalized polyhedral oligomeric silsesquioxanes (POSS), most interest has been drawn to hexahedral octasilsesquioxanes (T8) due to their high solubility in numerous organic solvents, allowing simple purification and making them useful building blocks for construction of a variety of hybrid materials with well-defined structure.7 Their complete functionalization can be for instance achieved by hydrosilation, Heck, and cross-metathesis approaches,8 and this field of synthetic chemistry has been the topic of some interesting reviews.9
The formation of higher SQ analogues such as T10 or T12 is the result of spontaneous rearrangement of T8 compounds and the formation of thermodynamically more stable cages.10 Such cage rearrangement has been reported by some research groups;11 however, there is little information available in the literature concerning pure, isolated larger cages like the aforementioned heptahedral T10. This fact arises inter alia from solubility differences between T8, T10, and T12 analogues. Especially, when both T10 and T12 compounds have similar chemical and physical properties, it is difficult to find a simple method of their separation.12 Moreover, separation of cage-like compounds possessing different numbers of side-chains is also cumbersome. For instance, Laine et al. pointed out that styrenyl-functionalized cage silsesquioxanes exhibited the same retention times during chromatographic purification, although they had different numbers of phenyl substituents.13 Because of that, it is crucial to realize that these species can simply be regarded as well-defined oligomers rather than discrete molecular compounds, even though the distinction may seem unnoticed.
The separation and purification of T8/T10/T12 mixtures are laborious and usually seem complex, especially taking into account the low yields of reactions and possibilities of cage rearrangements that lead to randomly structured silsesquioxanes (so-called T resins or organic silicates) such as oligomeric compounds, “aleatory” silicon-based species, open-like cages, etc.14 All this entails complicated pathways of separation. For example, Kawakami et al. reported the formation of T8, T10 and T12 cages (octa-, deca-, and dodeca-4-nitrophenyl cage silsesquioxanes) during hydrolysis of 4-substituted-phenyltriethoxysilane in the presence of tetrabutylammonium fluoride (TBAF).15 To obtain pure fractions, various crystallizations using acetonitrile/THF (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), hexane, and ethanol/hexane (v/v, 1:4) systems were carried out, and each fraction was purified by further recrystallization. Also, Laine et al., inspired by the Kawakami procedure, utilized it for T10/12 stilbenevinyl-silsesquioxane separation.16 In another interesting report, Ervithayasuporn described the isolation of methacrylate- and acrylate-functionalized T8, T10 and T12 SQs.17 To the best of our knowledge, there are no more convincing examples of separation of cage-like silsesquioxanes. Moreover, there are no examples of T resin purification protocols devoted to SQs containing alkyl side-chains. Such separations proved difficult due to alkyl derivatives' high solubility in most solvents.
:1), hexane, and ethanol/hexane (v/v, 1:4) systems were carried out, and each fraction was purified by further recrystallization. Also, Laine et al., inspired by the Kawakami procedure, utilized it for T10/12 stilbenevinyl-silsesquioxane separation.16 In another interesting report, Ervithayasuporn described the isolation of methacrylate- and acrylate-functionalized T8, T10 and T12 SQs.17 To the best of our knowledge, there are no more convincing examples of separation of cage-like silsesquioxanes. Moreover, there are no examples of T resin purification protocols devoted to SQs containing alkyl side-chains. Such separations proved difficult due to alkyl derivatives' high solubility in most solvents.
In this article, we report the synthesis, isolation and characterization of novel transparent non-fluorinated organic–inorganic hybrids based on octa and deca(3-decanamidopropyl)silsesquioxanes with hydrophobic properties.
Experimental section
Materials
All commercially available chemicals were of reagent-grade quality and were used without further purification. Solvents were treated as follows: hexanes and diethyl ether were distilled from Na; acetone and dichloromethane were distilled from P2O5; ethanol (anhydrous, J. T. Baker), methanol, and chloroform (HPLC grade, Aldrich) were used as received. Dimethylformamide (99.8% anhydrous, Aldrich) was dried over activated 4 Å molecular sieves prior to use. Dimethyl sulfoxide (99.6%, Aldrich), decanoyl chloride (98%, Aldrich), (3-aminopropyl)triethoxysilane (99%, Fluorochem), (3-aminopropyl)trimethoxysilane (99%, Fluorochem), trifluoromethanesulfonic acid (98%, Apollo Scientific), triethylamine (99.5%, Aldrich), HCl (36–38%, Avantor Performance Materials), and NaHCO3 (Avantor Performance Materials) were used without further purification unless stated otherwise. Chromium(III) acetylacetonate was prepared following the described procedure.18 Synthesis of octa and deca(3-decanamidopropyl)silsesquioxanes 4 and 5 was carried out under nitrogen gas flow, in a Schlenk line. Glassware was predried at 120 °C. The empty syringes were purged with nitrogen gas before being used for injection of the solvents and reagents.Characterization methods
1H NMR and 13C NMR spectra were recorded using a Bruker Avance 500 or a Bruker Avance III 600 spectrometer equipped with broadband inverse gradient probeheads. 1H NMR spectra were collected at 500.13 MHz using 12500 Hz spectral width, a relaxation delay of 1.0 s, a pulse width of 7°, 131 K data points, number of scans 16. Spectra were referred to the residual solvent signals (DMSO-d6 2.50 ppm, CDCl3 7.26 ppm) or TMS (0.00 ppm) as an internal reference. 13C NMR spectra were collected at 125.77 MHz using 29761.9 Hz spectral width, a relaxation delay of 2.0 s, a pulse width of 15°, 65 K data points, number of scans 1024, referred to solvent signals (DMSO-d6, 39.52, CDCl3 77.16 ppm). 29Si NMR spectra were recorded on a Bruker AMX-300 spectrometer using Wildmad PTFE–FEP (polytetra-fluoroethylene/fluorinated ethylene polypropylene copolymer) 5 mm tube liners and were collected at 59.62 MHz using 23810 Hz spectral width, a relaxation delay of 10.0 s, a pulse width of 13°, 65 K data points, number of scans 14000. Cr(acac)3 was added in a concentration of ∼10−2 mol L−1 as a shiftless relaxation agent. Chemical shifts were referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) (δ 1.316 ppm) or tetramethylsilane (TMS) (δ 0.00 ppm). Two-dimensional NMR spectra were recorded with 2048 data points in the t2 domain and up to 2048 points in the t1 domain, with a 1.0 s recovery delay. All 2D spectra were recorded with gradient selection. DOSY experiments were performed on a Bruker Avance III 600 spectrometer equipped with an Accustar z-axis gradient amplifier and an ATMA BBO probe with a z-axis gradient coil. All experiments were run without spinning to avoid convection. Fourier-transform infrared spectra (FTIR) were recorded on a Bruker Vertex 70 FTIR spectrometer. The FTIR sample chamber was flushed continuously with N2 prior to data acquisition in the range 4000–400 cm−1 with a precision of ±1 cm−1. Samples' spectra were recorded as KBr pellets. Optical grade, random cuttings of KBr were ground, with 1.0 wt% of the sample to be analyzed and pressed KBr pellets. Powder X-ray diffraction (PXRD) patterns of the dried powders were recorded on Bruker D8 ADVANCE diffractometer equipped with a copper lamp (λCuKα = 1.5406 Å) at 30 kV and 40 mA with a slit of 1/2°. Standard measurements were done for 2θ = 5–50° with a 2θ step of 0.016° and a counting time of 1.38 s. High resolution and accurate mass spectra were carried out on a Bruker microTOF-Q spectrometer (Bruker Daltonics, Bremen, Germany) equipped with an ESI source. Samples 1–3 were dissolved in methanol, samples 4–6 in chloroform. The experimental parameters were as follows: scan range 200–2500 m/z, drying gas nitrogen, temperature 200 °C, ion source voltage 4500 V, in-source collision energy 10 eV. The instrument operated in the positive ion mode and was calibrated externally with Tunemix mixture (Bruker Daltonics, Germany). Analyte solutions were introduced at a flow rate of 4.0 L min−1. Compass Data Analysis (Bruker Daltonics, Germany) software was used to determine the formulae of the compounds. The distance between the isotopic peaks allowed calculation of the charge of the analyzed ions. Elemental analyses (C, H and N) were performed using a Vario EL III element analyzer. Thin layer chromatography (TLC) was performed on Merck silica gel 60 F254 plates. Chromatograms were visualized using UV light (254 nm). For detection of amine, the chromatograms were first dipped in a 5% (w/v) solution of ninhydrin in 95% aqueous ethanol, and finally charred on a hot plate. Flash column chromatography was performed with Merck silica gel, grade 60, 230–400 mesh. Moisture in the organic solvents was measurement by Karl Fischer titration using Mettler Toledo DL 39 coulometer. Thermogravimetry and differential thermal analyses (TG-DTA) were recorded with a Setaram SETSYS 16/18 instrument. Samples for thermogravimetric characterization were placed in open alumina crucibles in a synthetic air (60% N2, 40% O2) or dinitrogen atmosphere. A heating rate of 10 °C min−1 was applied and all samples were studied between 30 and 1000 °C. The purity of all novel compounds was determined by combustion analysis, which confirmed that they were at least 99% pure. DSC data (30 to 250 °C, 5 °C min−1) were collected using a Perkin Elmer DSC 8500 calorimeter. UV/vis absorption spectra were recorded in the range of 200 to 800 nm wavelength at intervals of 0.5 nm by a Cary 50 Bio spectrophotometer. Data were processed using Origin 8.0. The images were obtained using an FEI Tecnai G2 F20 X-TWIN Transmission Electron Microscope equipped with a Penta FET EDX detector at an acceleration voltage of 200 kV. Colloidal solutions of nanoparticles with a concentration of about 5 mg mL−1 in methanol were drop-cast on a 3 mm holey carbon copper grid and dried under an IR lamp for 15 min. Contact angle measurements were conducted with a VCA Optima XE at ambient temperature (22–25 °C) and relative humidity (20–40%). A separate testing syringe was used for each test liquid to avoid cross-contamination. A liquid droplet of 5 μL was formed at the end of the syringe and carefully deposited onto the sample surface. The syringe was withdrawn and an image of the static contact angle was taken within 3 s of liquid deposition by a charge coupled device (CCD) camera. The contact angle was calculated by vendor-supplied software. The reported contact angle values are based on 5–8 repeats.
Syntheses
Method A. To (3-aminopropyl)triethoxysilane (2.34 mL, 2.21 g, 10.0 mmol), CF3SO3H (0.5 M aqueous solution, 40 mL, 20.0 mmol, 2.0 eq.) was added with stirring at room temperature. The resulting solution was stirred for 1 h and then heated to 50 °C in an open beaker until the solvent had completely evaporated (ca. 8 h). The crude product was heated at 100 °C for 2 h, then cooled down to room temperature and acetone (200 mL) was added. The precipitate was isolated by filtration, washed with acetone (3 × 10 mL), and dried in a vacuum oven (25 °C, 0.5 mbar) to give 2 as a white powder. Yield: 2.392 g, 92%. Elemental analysis calcd (%) for C32H72F24N8O36S8Si8 (2081.67): C 18.46, H 3.49, N 5.38, S 12.32. Found: C 18.41, H 3.45, N 5.35, S 12.34. FT-IR (cm−1, KBr pellets): νN–H = 3041 (s), δNH3 = 1615 (m), νC–N = 1507 (m), νC–F = 1267 (s), νSi–C = 1226 (m), νSi–O–Si = 1138 (s), νSO3 = 1030 (s), νC–S = 640 (s). HR-MS (ESI+, TOF, MeOH), m/z: 881.2902 [M + H − 8CF3SO3H]+ (calcd 881.2871), 441.1472 [M + 2H − 8CF3SO3H]2+ (calcd 441.1472), 294.4401 [M + 3H − 8CF3SO3H]3+ (calcd 294.4339). 1H NMR (500 MHz, DMSO-d6, 300 K): δ = 7.57 (s, 24H, NH3+), 2.75 (t, 3JHH = 7.2 Hz, 16H, CH2NH3+), 1.56 (m, 16H, SiCH2CH2CH2NH3+), 0.66 (t, 3JHH = 8.6 Hz, 16H, SiCH2). 13C{1H} NMR (126 MHz, DMSO-d6, 300 K): δ = 122.5 (q, 1JC–F = 317 Hz, CF3SO3−), 41.2 (s, SiCH2CH2CH2NH3+), 20.7 (s, SiCH2CH2CH2NH3+), 8.6 (s, SiCH2CH2CH2NH3+). 29Si{1H} NMR (59.6 MHz, DMSO-d6, 300 K): δ = −66.53 (s, T3). Temperature of decomposition to SiO2 (determining by TGA measurement, air), residue yield: 428 °C, 23.12% (calcd 23.09%).
Method B. The procedure was analogous to that described in Method A with the use of (3-aminopropyl)trimethoxysilane (1.75 mL, 1.79 g, 10.0 mmol) instead of (3-aminopropyl)triethoxysilane. Yield: 2.418 g, 93%. Elemental analysis calcd (%) for C32H72F24N8O36S8Si8 (2081.67): C 18.48, H 3.41, N 5.39, S 12.31. Found: C 18.52, H 3.52, N 5.27, S 12.45. HR-MS (ESI+, TOF, MeOH), m/z: 881.2892 [M + H − 8CF3SO3H]+ (calcd 881.2871).
Method A. To (3-aminopropyl)triethoxysilane (2.34 mL, 2.21 g, 10.0 mmol), CF3SO3H (0.5 M aqueous solution, 40 mL, 20.0 mmol, 2.0 eq.) was added with stirring at room temperature. The resulting solution was stirred for 1 h and then heated to ca. 50 °C in an open beaker until the solvent had completely evaporated (ca. 8 h). The crude product was heated at 100 °C for 2 h, then cooled down to room temperature and acetone (200 mL) was added. The mixture was then filtered off and clear solution was evaporated and dried under vacuum (25 °C, 0.5 mbar) giving white powder in 3% yield (0.076 g). Elemental analysis calcd (%) for C40H88F30N10O45S10Si10 (2600.62): C 18.48, H 3.41, N 5.39, S 12.31. Found: C 18.40, H 3.46, N 5.28, S 12.20. FT-IR (cm−1, KBr pellets): νN–H = 3041 (s), δNH3 = 1615 (m), νC–N = 1507 (m), νC–F = 1267 (s), νSi–C = 1226 (m), νSi–O–Si = 1138 (s), νSO3 = 1031 (s), νC–S = 640 (s). HR-MS (ESI+, TOF, MeOH), m/z: 1101.3570 [M + H – 10CF3SO3H]+ (calcd 1101.3570), 551.1876 [M + 2H − 10CF3SO3H]2+ (calcd 551.1821), 367.7912 [M + 3H − 10CF3SO3H]3+ (calcd 367.7905). 1H NMR (500 MHz, DMSO-d6, 300 K, shift data obtained from DOSY spectrum): δ = 7.52 (s, 30H, NH3+), 2.73 (t, 3JHH = 7.2 Hz, 20H, CH2NH3+), 1.49 (m, 20H, SiCH2CH2CH2NH3+), 0.59 (t, 3JHH = 8.6 Hz, 20H, SiCH2). 13C{1H} NMR (126 MHz, DMSO-d6, 300 K): δ = 122.5 (q, 1JC–F = 317 Hz, CF3SO3−), 41.1 (s, SiCH2CH2CH2NH3+), 20.6 (s, SiCH2CH2CH2NH3+), 8.2 (s, SiCH2CH2CH2NH3+). 29Si{1H} NMR (59.6 MHz, DMSO-d6, 300 K): δ = −68.55 (s, T3). Temperature of decomposition to SiO2 (determining by TGA measurement, air), residue yield: 415 °C, 23.00% (calcd 23.09%).
Method B. The procedure was analogous to that described in Method A with the use of (3-aminopropyl)trimethoxysilane (1.75 mL, 1.79 g, 10.0 mmol) instead of (3-aminopropyl)triethoxysilane. Yield: 0.016 g, 0.5%. Elemental analysis calcd (%) for C40H88F30N10O45S10Si10 (2600.62): C 18.48, H 3.41, N 5.39, S 12.31. Found: C 18.52, H 3.36, N 5.44, S 12.33. HR-MS (ESI+, TOF, MeOH), m/z: 367.7963 [M + 3H − 10CF3SO3H]3+ (calcd 367.7905). 29Si{1H} NMR (59.6 MHz, DMSO-d6, 300 K): δ = −68.55 (s, T3).
Method C. CF3SO3H (0.091 mL, 0.154 g, 1.02 mmol, 12 eq.) was added dropwise to a solution of 1 (0.100 g, 0.0852 mmol) in DMSO (5 mL). The resulting solution was stirred for 2 h at 40 °C, then cooled down to room temperature, then DMSO was blown out with N2 to give yellowish solid. T10 SQ was extracted by washing with acetone (3 × 15 mL). Next, acetone was evaporated under reduced pressure. The analytically pure product can be obtained as a white microcrystalline powder by recrystallization from MeOH to give a white powder in 44% yield (0.078 g). Elemental analysis calcd (%) for C40H88F30N10O45S10Si10 (2600.62): C 18.48, H 3.41, N 5.39, S 12.31. Found: C 18.71, H 3.31, N 5.32, S 12.62. HR-MS (ESI+, TOF, MeOH), m/z: 1101.3612 [M + H − 10CF3SO3H]+ (calcd 1101.3570), 567.7398 [M + 3H − 6CF3SO3H]3+ (calcd 567.7367), 517.7479 [M + 3H − 7CF3SO3H]3+ (calcd 517.7504), 467.7697 [M + 3H − 8CF3SO3H]3+ (calcd 467.7638), 426.1133 [M + 4H − 6CF3SO3H]4+ (calcd 426.0545), 417.7837 [M + 3H − 9CF3SO3H]3+ (calcd 417.7771), 401.4678 [M + 5H − 4CF3SO3H]5+ (calcd 401.2029), 388.5721 [M + 5H − 7CF3SO3H]5+ (calcd 388.5646), 371.7947 [M + 5H − 5CF3SO3H]5+ (calcd 371.0371), 367.8035 [M + 5H − 10CF3SO3H]5+ (calcd 367.7905), 351.0974 [M + 4H − 8CF3SO3H]4+ (calcd 351.0746), 313.5899 [M + 4H − 9CF3SO3H]4+ (calcd 313.5847), 311.0972 [M + 5H − 7CF3SO3H]5+ (calcd 311.0531), 281.0678 [M + 5H − 8CF3SO3H]5+ (calcd 281.0612), 276.0995 [M + 4H − 10CF3SO3H]4+ (calcd 276.0947), 250.8448 [M + 5H − 9CF3SO3H]5+ (calcd 250.8676). 29Si{1H} NMR (59.6 MHz, DMSO-d6, 300 K): δ = −68.55 (s, T3).
Method D. The procedure was analogous to that described in Method C with the use 2 (0.177 g, 0.0852 mmol) instead of 1. Yield: 0.122 g, 55%. Elemental analysis calcd (%) for C40H88F30N10O45S10Si10 (2600.62): C 18.48, H 3.41, N 5.39, S 12.31. Found: C 18.51, H 3.48, N 5.35, S 12.26. HR-MS (ESI+, TOF, MeOH), m/z: 367.8000 [M + 3H − 10CF3SO3H]3+ (calcd 367.7905). 29Si{1H} NMR (59.6 MHz, DMSO-d6, 300 K): δ = −68.55 (s, T3).
Method A. Synthesis of 4 was carried out under a N2 atmosphere. Decanoyl chloride (1.46 mL, 1.31 g, 6.89 mmol, 8.08 eq.) was added dropwise to a suspension of 1 (1.000 g, 0.852 mmol) and NEt3 (2.10 mL, 1.53 g, 15.1 mmol, 17.7 eq.) in DMF (40 mL, 0 °C). After overnight stirring, the crude product was precipitated by slow addition of 1 M aqueous HCl (70 mL, 0 °C). Filtration, washing with cold saturated NaHCO3 (70 mL), next with water and then drying under vacuum (25 °C, 0.5 mbar) afforded a white solid of 4 (yield 1
:1.280 g, 71%). The residue was purified by flash column chromatography (Et2O/hexane, v/v, 1:1) to obtain 4 as a white solid in 70% (yield 2:1.265 g). Rf = 0.56 (Et2O/hexane, v/v, 1:1). Elemental analysis calcd (%) for C104H208N8O20Si8 (2115.49): C 59.05, H 9.91, N 5.30. Found: C 59.09, H 9.87, N 5.32. FT-IR (cm−1, KBr pellets): νN–H = 3278 (s), νC–H = 2931 (m), νC–H = 2871 (m), νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1636 (s), δNH = 1558 (s), νC–N = 1457 (w), νring-asymSi–O–Si = 1122 (s), δO–Si–O = 698 (w), δSi–O–Si = 472 (w). HR-MS (ESI+, TOF, CHCl3), m/z: 2114.3735 [M + H]+ (calcd 2114.3732), 2015.3122 [M − C7H14 + H]+ (calcd 2015.2558), 1057.6976 [M + 2H]2+ (calcd 1057.6902), 705.4905 [M + 3H]3+ (calcd 705.4626). 1H NMR (500 MHz, CDCl3, 300 K): δ = 3.12 (t, 3JHH = 7.0 Hz, 16H, CH2NH), 2.47 (t, 3JHH = 7.4 Hz, 16H, C(O)CH2), 1.50 (br, 32H, SiCH2CH2CH2 and C(O)CH2CH2), 1.22–1.25 (br, 96H, –CH2–), 0.79 (t, 3JHH = 7.0 Hz, 24H, CH3), 0.52 (t, 3JHH = 8.4 Hz, 16H, SiCH2). 13C{1H} NMR (126 MHz, CDCl3, 300 K): δ = 169.3 (s, CO), 42.4 (s, SiCH2CH2CH2NH), 36.8 (s, C(O)CH2), 32.0, 29.6, 29.5, 29.4, 29.4, 29.3 (s, –CH2–), 23.5 (s, SiCH2CH2CH2NH), 22.8 (s, C(O)CH2CH2), 14.3 (s, CH3), 9.3 (s, SiCH2CH2CH2NH). 29Si{1H} NMR (59.6 MHz, CDCl3, 300 K): δ = −66.33 (s, T3). Temperature of decomposition to SiO2 (determined by TGA measurement, air), residue yield: 450 °C, 22.72% (calcd 22.76%).
O = 1636 (s), δNH = 1558 (s), νC–N = 1457 (w), νring-asymSi–O–Si = 1122 (s), δO–Si–O = 698 (w), δSi–O–Si = 472 (w). HR-MS (ESI+, TOF, CHCl3), m/z: 2114.3735 [M + H]+ (calcd 2114.3732), 2015.3122 [M − C7H14 + H]+ (calcd 2015.2558), 1057.6976 [M + 2H]2+ (calcd 1057.6902), 705.4905 [M + 3H]3+ (calcd 705.4626). 1H NMR (500 MHz, CDCl3, 300 K): δ = 3.12 (t, 3JHH = 7.0 Hz, 16H, CH2NH), 2.47 (t, 3JHH = 7.4 Hz, 16H, C(O)CH2), 1.50 (br, 32H, SiCH2CH2CH2 and C(O)CH2CH2), 1.22–1.25 (br, 96H, –CH2–), 0.79 (t, 3JHH = 7.0 Hz, 24H, CH3), 0.52 (t, 3JHH = 8.4 Hz, 16H, SiCH2). 13C{1H} NMR (126 MHz, CDCl3, 300 K): δ = 169.3 (s, CO), 42.4 (s, SiCH2CH2CH2NH), 36.8 (s, C(O)CH2), 32.0, 29.6, 29.5, 29.4, 29.4, 29.3 (s, –CH2–), 23.5 (s, SiCH2CH2CH2NH), 22.8 (s, C(O)CH2CH2), 14.3 (s, CH3), 9.3 (s, SiCH2CH2CH2NH). 29Si{1H} NMR (59.6 MHz, CDCl3, 300 K): δ = −66.33 (s, T3). Temperature of decomposition to SiO2 (determined by TGA measurement, air), residue yield: 450 °C, 22.72% (calcd 22.76%).
Method B. Synthesis performed under a N2 atmosphere. An excess of decanoyl chloride (1.63 mL, 1.47 g, 7.69 mmol, 16.0 eq.) was added dropwise to a solution of 2 (1.00 g, 0.480 mmol) and NEt3 (1.77 mL, 1.28 g, 8.46 mmol, 17.6 eq.) in DMF (40 mL, 0 °C). After overnight stirring, the crude product was precipitated by slow addition to 1 M aqueous HCl (40 mL, 0 °C). Filtration, washing with cold saturated NaHCO3 (70 mL), next with water and then drying in vacuo (25 °C, 0.5 mbar) afforded a white solid. The residue was purified by flash column chromatography (Et2O/hexane, v/v, 1
:1) to afford 4 as a white solid in 86% (yield: 0.870 g). Rf = 0.56 (Et2O/hexane, v/v, 1:1). Elemental analysis calcd (%) for C104H208N8O20Si8 (2115.49): C 59.05, H 9.91, N 5.30. Found: C 59.10, H 9.86, N 5.35. FT-IR (cm−1, KBr pellets): νN–H = 3278 (s), νC–H = 2931 (m), νC–H = 2871 (m), νCO = 1636 (s), δNH = 1558 (s), νC–N = 1457 (w), νring-asymSi–O–Si = 1122 (s), δO–Si–O = 698 (w), δSi–O–Si = 472 (w). HR-MS (ESI+, TOF, CHCl3), m/z: 2114.3742 [M + H]+ (calcd 2114.3732), 1057.6976 [M + 2H]2+ (calcd 1057.6902). 1H NMR (500 MHz, CDCl3, 300 K): δ = 3.12 (t, 3JHH = 7.0 Hz, 16H, CH2NH), 2.47 (t, 3JHH = 7.4 Hz, 16H, C(O)CH2), 1.50 (br, 32H, SiCH2CH2CH2) and C(O)CH2CH2, 1.22–1.25 (br, 96H, –CH2–), 0.79 (t, 3JHH = 7.0 Hz, 24H, CH3), 0.52 (t, 3JHH = 8.4 Hz, 16H, SiCH2). 13C{1H} NMR (126 MHz, CDCl3, 300 K): δ = 169.2 (s, CO), 42.2 (s, SiCH2CH2CH2NH), 36.8 (s, C(O)CH2), 32.0, 29.6, 29.5, 29.4, 29.3, (s, –CH2–), 23.0 (s, SiCH2CH2CH2NH), 22.7 (s, C(O)CH2 CH2), 14.1 (s, CH3), 9.2 (s, SiCH2CH2CH2NH). 29Si{1H} NMR (59.6 MHz, CDCl3, 300 K): δ = −66.32 (s, T3). Temperature of decomposition to SiO2 (determined by TGA measurement, air), residue yield: 450 °C, 22.76% (calcd 22.72%).
Method A. Synthesis of 5 was carried out under a N2 atmosphere. Decanoyl chloride (0.081 mL, 0.074 g, 0.388 mmol, 10.1 eq.) was added dropwise to a solution of 3 (0.100 g, 0.0385 mmol) and NEt3 (0.119 mL, 0.086 g, 0.850 mmol, 22.1 eq.) in DMF (4 mL, 0 °C). After overnight stirring, the crude product was precipitated by slow addition of 1 M aqueous HCl (10 mL, 0 °C). Obtained precipitate was filtered off and washed with cold saturated NaHCO3 (10 mL) and water and then dried under vacuum (25 °C, 0.5 mbar) afforded a white solid of 5. The residue was purified by flash column chromatography (Et2O/hexane, v/v, 1
:1) to afford 5 as a white solid in 81% (yield: 0.082 g). Rf = 0.40 (Et2O/hexane, v/v, 1:1). Elemental analysis calcd (%) for C130H260N10O25Si10 (2644.45): C 59.05, H 9.91, N 5.30. Found: C 59.01, H 9.93, N 5.29. FT-IR (cm−1, KBr pellets): νN–H = 3278 (s), νC–H = 2931 (m), νC–H = 2871 (m), νCO = 1636 (s), δNH = 1558 (s), νC–N = 1457 (w), νring-asymSi–O–Si = 1122 (s), δO–Si–O = 698 (w), δSi–O–Si = 472 (w). HR-MS (ESI+, TOF, CHCl3), m/z: 1321.8756 [M + 2H]2+ (calcd 1321.8610), 881.5648 [M + 3H]3+ (calcd 881.5764). 1H NMR (500 MHz, CDCl3, 300 K): δ = 3.15 (t, 3JHH = 7.0 Hz, 20H, CH2NH), 2.51 (t, 3JHH = 7.4 Hz, 20H, C(O)CH2), 1.52 (br, 40H, SiCH2CH2CH2 and C(O)CH2 CH2, 1.20–1.25 (br, 120H, –CH2–), 0.82 (t, 3JHH = 7.0 Hz, 30H, CH3), 0.56 (t, 3JHH = 8.4 Hz, 20H, SiCH2). 13C{1H} NMR (126 MHz, CDCl3, 300 K): δ = 169.3 (s, CO), 42.2 (s, SiCH2CH2CH2NH), 31.9 (s, C(O)CH2), 30.7, 29.5, 29.4, 29.3, 29.3, 29.2, (s, –CH2–), 22.9 (s, SiCH2CH2CH2NH), 22.7 (s, C(O)CH2CH2), 14.1 (s, CH3), 9.2 (s, SiCH2CH2CH2NH). 29Si{1H} NMR (59.6 MHz, CDCl3, 300 K): δ = −68.54 (s, T3). Temperature of decomposition to SiO2 (determined by TGA measurement, air), residue yield: 417 °C, 22.74% (calcd 22.72%).
Method B. Synthesis performed under a N2 atmosphere. An excess of decanoyl chloride (1.63 mL, 1.47 g, 7.69 mol) was added dropwise to a solution of mixture 2 and 3, obtained during reaction of APTES and CF3SO3H before isolation of T8 and T10 compounds (1.00 g), and NEt3 (1.77 mL, 1.284 g, 12.67 mmol) in DMF (40 mL, 0 °C). After overnight stirring, the crude product was precipitated by slow addition to 1 M aqueous HCl (40 mL, 0 °C). Filtration, washing with cold saturated NaHCO3 (70 mL), next with water and then drying in vacuo (25 °C, 0.5 mbar) afforded a white solid. The residue was purified by flash column chromatography (Et2O/hexane, v/v, 1
:1) to afford 5 as a white solid in 2% (yield: 0.029 g). Rf = 0.40 (Et2O/hexane, v/v, 1:1). Elemental analysis calcd (%) for C130H260N10O25Si10 (2644.45): C 59.05, H 9.91, N 5.30. Found: C 59.07, H 9.87, N 5.27. HR-MS (ESI+, TOF, CHCl3), m/z: 1321.8656 [M + 2H]2+ (calcd 1321.8610), 881.5782 [M + 3H]3+ (calcd 881.5764), 661.4322 [M + 4H]4+ (calcd 661.4341). 29Si{1H} NMR (59.6 MHz, CDCl3, 300 K): δ = −68.54 (s, T3).
O), 41.2, 36.9, 31.8, 29.5, 29.4, 29.4, 29.3, 26.0, 22.9, 22.7 (s, –CH2–), 14.1, 11.4 (s, –CH3). Temperature of decomposition (determined by TGA measurement, air), residue yield: 265 °C, 0.05% (calcd 0.00%).Results and discussion
Synthesis, separation and characterization of 1–6
The development of both high-yield syntheses and effective separations that provide access to pure cage-like alkyl derivatives of silsesquioxanes has been long sought. In this work we focused our attention on T10 silsesquioxanes containing 3-decanamidopropyl side-chains and their isolation from the T8-like SQ analogue. In the first thrust, we obtained three crystalline amino-SQ salts: octa(3-aminopropyl)silsesquioxane hydrochloride (1), octa(3-aminopropyl)silsesquioxane trifluoromethanesulfonate (2) and deca(3-aminopropyl)silsesquioxane trifluoromethanesulfonate (3). Compound 1 is a T8-type SQ and was prepared with the previously reported procedure18 based on studies by Feher19 and Laine20 (see also Wacker-Chemie's US patent21) by using commercially available (3-aminopropyl)triethoxysilane (APTES) and 3.6 equiv. of hydrochloric acid and its purity was analytically confirmed by 1H, 13C, 29Si NMR, FTIR, HR-MS, EDS, and PXRD (see ESI Fig. S3–S9†). Octa(3-aminopropyl)silsesquioxane trifluoromethanesulfonate was also prepared by Kaneko et al. by using (3-aminopropyl)trimethoxysilane (APTMS) but without observation of decamer formation.22 Here the mixture of T8 SQ 2 and T10-type SQ 3 was prepared in the reaction of APTES (or (3-aminopropyl)trimethoxysilane, APTMS) with 0.5 M trifluoromethanesulfonic acid (CF3SO3H) in 1:2 molar ratio. To extract 3 from the crude product, the resulting powder was washed with acetone to provide a pure crystalline powder of 2 (92% yield), and T10 cage-like 3 was isolated from the resulting acetone solution by simple evaporation as a white powder in 3% yield (In the ‘Cage-rearrangement’ paragraph we precisely described a Method C that allowed us to obtain T10-cage with a higher yield in response to the T8 core's reorganization). The high purity of 2 and 3 was unambiguously confirmed with high-resolution mass spectrometry and multinuclear (1H, 13C, 29Si) NMR spectroscopy. Furthermore, powder X-ray diffraction PXRD measurements were performed (see ESI Fig. S10–S30†). Ervithayasuporn et al.23 demonstrated that SQs' physical properties are closely related to their core symmetry. Highly symmetrical T8s form crystalline compounds, whereas T10 and T12, with lower symmetry D5h and D2d, respectively, tend to have a loosely packed structure that can be postulated by powder XRD23 and manifested by the appearance of broad diffraction peaks, instead of sharp, as shown in Fig. 1 (see also ESI Fig. S1, S16 and S29†). In the case of 3 (T10), observed broadening may indicate irregular packing in the solid state.
 | ||
| Fig. 1 Powder XRD spectra of 2 (T8) and 3 (T10). | ||
The 29Si chemical shifts of 2 (T8) and 3 (T10) appeared at −66.5 and −68.6 ppm (Fig. 2, see also ESI Fig. S12, S19–S22†), respectively, which are within the expected regions for alkyl-substituted cage-SQs.24
 | ||
| Fig. 2 29Si NMR spectra of 1, 2, 3 and 1, 2 after reaction with CH3SO3H. | ||
The heptahedral structure of 3 was also studied by proton NMR. Due to the fact that 1H NMR spectra of 2 and 3 are identical (proton chemical shifts characteristic for –NH3+, –CH2NH3+, –SiCH2CH2CH2NH3+ and –SiCH2– fragments are shown at 7.52, 2.73, 1.49, and 0.59 ppm, respectively), we performed sophisticated DOSY (diffusion ordered spectroscopy) experiments as shown in Fig. 3 (see also ESI Fig. S30†), which allowed differentiation of the compounds with different mass and symmetry.
 | ||
| Fig. 3 1H DOSY (600 MHz, DMSO-d6, 300 K) spectrum of mixtures 2 and 3. | ||
Determined diffusion coefficients equal 6.18 × 10−7 and 6.80 × 10−7 cm2 s−1 for 2 and 3, respectively. In the case of 3, the higher diffusion coefficient factor derived from its higher mass. Here, motion of the particles is slighter, which strongly affects the lower diffusion in DMSO-d6 solution.
In turn, 13C NMR spectra of 2 and 3 differ, and these compounds can be clearly distinguished. For 2 carbon chemical shifts are 122.5 (CF3SO3−), 41.2 (s, SiCH2CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H2NH3+), 20.7 (s, SiCH2H2CH2NH3+), 8.6 (s, SiH2CH2CH2NH3+) ppm, and for 3 122.5 (CF3SO3−), 41.1 (s, SiCH2CH2H2NH3+), 20.6 (s, SiCH2H2CH2NH3+), 8.2 (s, SiH2CH2CH2NH3+) ppm (Fig. 4).
H2NH3+), 20.7 (s, SiCH2H2CH2NH3+), 8.6 (s, SiH2CH2CH2NH3+) ppm, and for 3 122.5 (CF3SO3−), 41.1 (s, SiCH2CH2H2NH3+), 20.6 (s, SiCH2H2CH2NH3+), 8.2 (s, SiH2CH2CH2NH3+) ppm (Fig. 4).
 | ||
| Fig. 4 13C NMR spectra of 2 (top) and 3 (bottom). | ||
In the case of 3, carbon signals are shifted upfield, and a slight difference is observed for those carbon atoms that are directly bonded to silicon atoms that form the siloxane core (note that the same relation is later observed for amide T8 and T10 compounds).
A similar phenomenon exists in the 29Si NMR spectra. It is worth noting here that in this field of studies, 13C NMR experiments are unfairly marginalized in the literature and authors mainly focus on silicon spectra, analyzing products of cage rearrangements and products of condensation reactions. We strongly believe that our analyses can be helpful in the future in interpreting octa- and decameric SQs based on 13C NMR.
In the infrared spectra of amine-SQs (ESI Fig. S6, S13 and S23†) specific vibration bands for terminal groups are indicated as broad peaks localized at 3041 (νN–H) and 1615 (δNH3) cm−1. Vibrations at 2935 (νC–H/sym) and 2872 (νC–H/asym) cm−1 correspond to methylene groups. In turn, vibration of the Si–C bond is observed at 798 cm−1. Moreover, the trifluoromethanesulfonate anion gave specific absorption bands at 1267, 1030 and 640 cm−1 and can be clearly assigned to ν(C–F), ν(SO3), and ν(C–S) modes, respectively. In the case of 1 and 2, the absorption band in the range of siloxane Si–O–Si bond vibrations is narrow and strong, and appears at 1116 and 1138 cm−1 for 1 and 2, respectively. In the case of 3, this vibration is broad and localized in the range of 1148 and 1085 cm−1.
Recently, we developed a synthetic strategy to produce large quantities of amido-functionalized polyhedral oligomeric silsesquioxanes by using acyl chlorides. The resulting compounds 1–3 were excellent candidates for subsequent transformation of this type. Reaction of 1 with decanoyl chloride and triethylamine (NEt3) in dimethylformamide (DMF) under dinitrogen resulted in octa(3-decanamidopropyl)silsesquioxane (4) in 70% yield (Scheme 1).
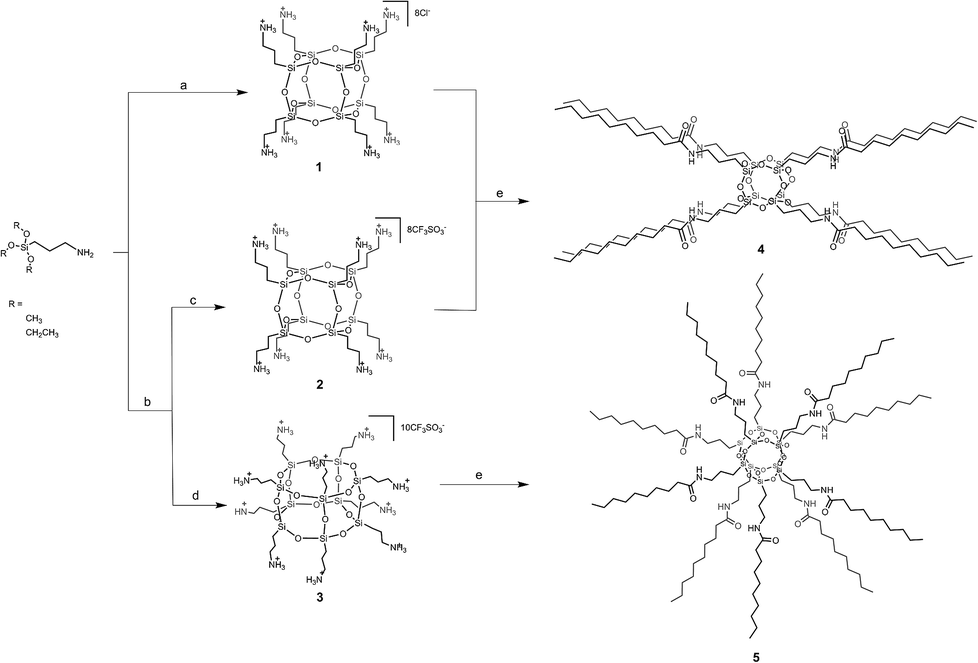 | ||
| Scheme 1 Synthesis of 1–5. (a) H2O, HCl; (b) H2O, CF3SO3H (c) acetone, precipitate (d) acetone solution (e) decanoyl chloride, DMF, NEt3. | ||
This conversion smoothly proceeded without any cage rearrangement. The 29Si NMR (ESI Fig. S35†) strongly indicated the presence of only one type of silicon nuclei expected for a cage-like structure and exhibited a single resonance at −66.3 ppm. The mass spectrum also confirmed the formation of a closed frame structure composed of eight Si atoms that bind decanamidopropyl side-chains {HRMS (ESI+, TOF, CHCl3): m/z: 2114.3735 [M + H]+ (calcd 2114.3732), 2015.3122, 1057.6976 [M + 2H]2+ (calcd 1057.6902), 705.4905 [M + 3H]3+ (calcd 705.4626)}.
DOSY NMR experiments (ESI Fig. S34 and S42†) of amido-functionalized compounds 4 and 5 resulted in diffusion coefficients (measured in chloroform) 2.96 × 10−6 and 5.76 × 10−6 cm2 s−1 for 4 (T8) and 5 (T10), respectively. Similarly to values observed for 2 and 3, amido derivative 5 with higher mass and lower symmetry has a higher diffusion coefficient.
In the FT-IR spectra (Fig. 5) of amido derivatives, vibration bands characteristic for amide groups are observed at 3278, 1636, 1558 and 1457 cm−1 and can be assigned to ν(N–H), ν(CO), δ(NH) and ν(C–N), respectively. Due to the presence of long alkyl chains, strong bands of methylene groups are observed at 2931 and 2871 cm−1, and also vibrations assigned to C–C bonds in the ‘fingerprint’ range (for full range spectra see ESI Fig. S36 and S44†). In the case of amido-functionalized compounds, vibration bands of siloxane modes are also observed. The octamer possesses a narrow strong band at 1122 cm−1, whereas for the decamer there is a broad band in the range of 1152 and 1090 cm−1. Moreover, the substitution of all R side-groups is clearly confirmed by the lack of vibration bands characteristic for amine groups and trifluoromethanesulfonate anions.
 | ||
| Fig. 5 FT-IR spectra of 2–5 (KBr pellets). | ||
Similarly, the reaction of the mixture of 2 and 3 with excess decanoyl chloride and NEt3 in DMF resulted in the formation of hexa 4 and heptahedral 5 species which were easily separated by flash column chromatography using Et2O/hexane (v/v, 1:1) as an eluent. 29Si NMR of 5 (ESI Fig. S43†) showed a singlet at −68.5 ppm. The Si chemical shift of 5, compared to that observed for 4 (Fig. S35†), is about 2.21 ppm shifted upfield as a result of the presence of eight- and ten-membered rings. Moreover, the compositions of 4 and 5 were confirmed by high resolution mass spectrometry and spectroscopic studies (see ESI Fig. S37, S45 and S46†). It should be noted that 4 and 5 can be separately obtained from 2 and 3 (see Experimental section), respectively. Nevertheless, since the final mixture of 4 and 5 can be more readily isolated by column chromatography, we performed the reaction as mentioned to facilitate the process (no need to conduct two independent reactions).
Cage-rearrangement
We also performed an experiment at the NMR scale to explain the catalyst's influence on the formation of larger cages. To a solution of 1 in DMSO-d6 14 eq. of trifluoromethanesulfonic acid CF3SO3H were added (8 eq. for anion exchange, and 6 eq. for reaction with siloxane core). During its addition, rapid gas liberation was observed. In the 29Si NMR spectrum (Fig. 2, see ‘1 + CF3SO3H’ spectrum) chemical shifts expected for cage rearrangement were noted that clearly confirmed the presence of a T10-like compound. In our opinion, this proves that the superacid catalyst has a decisive influence on the formation of silsesquioxane decamer 3 (Scheme 2). | ||
| Scheme 2 Direct synthesis of 3 (T10) from 1 using superacid CF3SO3H. | ||
In addition, we performed an experiment where we obtained and isolated 3 in 44% yield (see Method C for 1 → 3 transformation in Experimental section. It should be noted that it is also possible to obtain 3 starting from 2 in the same conditions. Here, the yield is even higher and equals 55% – see Method D in Experimental section and Fig. S22 and S27 in ESI†). In this case, reaction of hexahedral 1 and CF3SO3H in DMSO conducted in 1:12 molar ratio. Moreover, in order to trace the path of this reaction we have made several attempts in various stoichiometric ratios of 1:CF3SO3H = 1:1, 1:4, 1:8, 1:12, and 1:16 and reactions were monitored by 29Si NMR (Fig. S61†). It should be noticed that after addition of merely 1 eq. of acid, CF3SO3H is attached to silicon atoms. We isolated intermediates and examined them by using HR-MS and silicon NMR (Fig. S62–S64†). Based on these studies and after careful analyses we assumed that in the first step CF3SO3H acid attacks siloxane Si–O–Si bonds and the formation of Si–O–SO2CF3 bond parallel with cage opening process is observed and compound B is obtained (Fig. 6). Such an inversion is observed at silicon atom during nucleophilic displacement reaction that is usually noticed when leaving groups are replaced by soft nucleophiles.25 Upon further acid attack, both T6(OH)4 C and siloxane dimer D are formed. Because this reaction takes place in an aqueous conditions, compound E of general formula T8(OH)4 as a consequence of hydrolysis reaction was obtained. E is prone to reaction with D and due to this, the abstraction of CF3SO3− anion occurs and the closure frame with the spontaneous cage-rearrangement to heptahedral T10 structure F is observed. Although, heptahedral F is less favorable energetically (see below MM2 data, Fig. 7), in this case its creation is forces by the formation of a new Si4O4 moiety from much more less stable substrates D and E.
 | ||
| Fig. 6 Top: 29Si NMR spectra of 1 → 3 cage-rearrangement. Bottom: Reaction of OAS-POSS-Cl with CF3SO3H in DMSO containing water. B–E constitute intermediates isolated during A (1) → F (3) cage-rearrangement. | ||
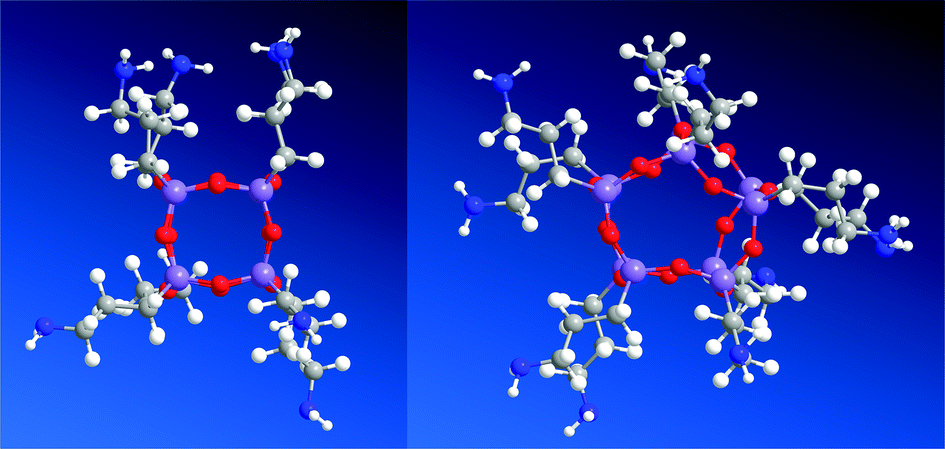 | ||
| Fig. 7 Molecular models of hexahedral 2 (left), heptahedral 3 (right). | ||
MM2 simulations of the obtained hexahedral 2 and heptahedral 3 are depicted in Fig. 7. The total energy of 2 is 11.72 kcal mol−1 and 31.10 kcal mol−1 for its amide derivative 4, which was apparently lower than that of 3 silsesquioxane 60.92 kcal mol−1 and 102.35 kcal mol−1 for amide-5. The simulations results were collected in Table S1.†
TG-DTA studies
The use of organic–inorganic hybrids constitutes one of the best solutions for materials to enhance their thermal and mechanical properties compared to those based on conventional organic systems.26 The thermal properties of 1–6 were characterized by thermal gravimetric analysis (TGA-DTA) performed under air and dinitrogen and by differential scanning calorimetry (DSC) under anaerobic conditions (N2). These data are summarized in Table 1 (see also ESI Fig. S51–S57†).| Compound | Temperature of extrusion of side chains [°C] | Temperature of decomposition [°C] | Calculated residue yield [%] | Residue yield [%] | Crystallization temperature [°C] | ΔH [kJ mol−1] | Melting temperature [°C] | ΔH [kJ mol−1] | |
|---|---|---|---|---|---|---|---|---|---|
| a Decomposition to silicon dioxide SiO2.b Decomposition to carbon residue. | |||||||||
| Air | 1 | — | 314a | 40.97 | 40.70 | — | — | — | — |
| 2 | 397 | 428a | 23.09 | 23.00 | — | — | — | — | |
| 3 | — | 414a | 23.11 | 23.32 | — | — | — | — | |
| 4 | 350 | 471a | 22.72 | 22.75 | — | — | — | — | |
| 5 | 312 | 451a | 22.72 | 22.74 | — | — | — | — | |
| 6 | — | 265b | 0.00 | 0.05 | — | — | — | — | |
| N2 | 4 | 374 | 454 | 22.72 | 23.08 | 178.24 | −27.90 | 189.51 | 40.98 |
| 5 | 398 | 440 | 22.72 | 28.81 | 183.56 | −48.20 | 192.74 | 87.37 | |
| 6 | — | 277 | 0.00 | 0.25 | 33.89 | −20.11 | 41.95 | 19.08 | |
Decomposition temperatures of salts 1–3 ranges from 314 to 428 °C. Octa 4 and decamer 5 SQs decompose at 471 and 451 °C, respectively. Compounds 4 and 5 possess higher thermal stability compared to that observed for 1–3. This phenomenon derives from the fact that amide species in comparison to amine-like compounds are much more stable at higher temperatures. T10-like compounds 3 and 5 are less stable than T8. For better stability of octa-cages response thermodynamically stable four Si4O4 moieties. Thermal decomposition (under air conditions) of amino-silsesquioxanes 1–3 proceeds in two steps. In the first step terminal groups are oxidized and in the TG curve a significant weight loss is observed. At the same time, the exoenergetic DTA peak is associated with heat evolution accompanied by oxidation of side chains. In the second step, the POSS core (SiO1.5)n (where n = 8, 10) decomposes to amorphous silica (SiO2)n. This process is associated with addition of an oxygen atom with concomitant abstraction of organic moieties. It should be noted that to fully understand decomposition process TG-MS technique should be employed. In this studies no TG-MS analyses were performed. After thermal decomposition, resulting products were examined using powder XRD (see ESI Fig. S2†) and FT-IR, and the obtained results clearly confirmed the formation of silicon dioxide. Moreover, to emphasize the advantage of alkyl amido-functionalized SQs over pure organic compounds, we additionally synthesized the N-propyldecanamide derivative (6) and performed its thermal analysis under similar conditions, which showed 6 to decompose at 265 °C, which is a dramatically lower temperature in comparison to hybrid species 4 and 5.
For amide compounds TG-DTA experiments were also performed in air (60% N2, 40% O2) and additionally in a nitrogen (N2) atmosphere. TG-DTA curves (air) for 4 and 5 differ from those observed for 1–3. Here, thermal decomposition is characterized by three distinct steps that can be assigned to amide bond cleavage, oxidation of terminal groups and destruction of siloxane core and final formation of silica. In the case of experiments performed in nitrogen, only two significant thermolysis steps are observed. These decompositions occur at higher temperatures compared to air conditions and are related to the lack of oxidizing conditions and processes that were mentioned above. In turn, temperatures of the decompositions are lower, and this can be assigned to the lack of oxygen “incorporation” into the siloxane (SiO1.5)n core. Based on DTA curves (dinitrogen atmosphere) melting temperatures for amide-SQs 4 and 5 can be designated. Their values clearly correspond to those observed in DSC (Table 1). For experiments performed in air conditions no melting points were noted, due to the fact that melting is associated with concomitant decomposition. As a comparison, pure organic 6 (without siloxane core) melts at 42 °C and starts to decompose at 178 °C; at 265 °C N-propyldecanamide achieves the highest rate of decomposition (Fig. 8).
 | ||
| Fig. 8 TG (black line), thermogram of 5 and 6 10 °C min−1 (in the air atmosphere: 60% N2, 40% O2). | ||
DSC studies
Compounds 1–3 melt and decompose in parallel, as was first observed in TG-DTA studies. In turn, amide derivatives 4 and 5 melt before decomposition and their melting temperatures are 190 and 193 °C, respectively. The attachment of amide fragments to the siloxane core causes an increase not only of melting points but also decomposition temperatures. Such phenomena have a practical aspect, not only from the hydrophobic surface point of view but such materials also constitute perfect covers in industrial applications when higher temperatures are used. Silsesquioxanes that possess long alkyl chains exhibit lower melting temperatures than derivatives containing an additional amide fragment. For instance, T8-C10H21 melts at 63 °C.27Melting and freezing temperatures of 4–6 determined by DSC are shown in Table 1, and their thermograms can be found in ESI (Fig. S58–S59†).
During heating/freezing DSC cycle compound 4 possesses outsized phase transition that is not observed in the case of 5 and 6 (Fig. 9).
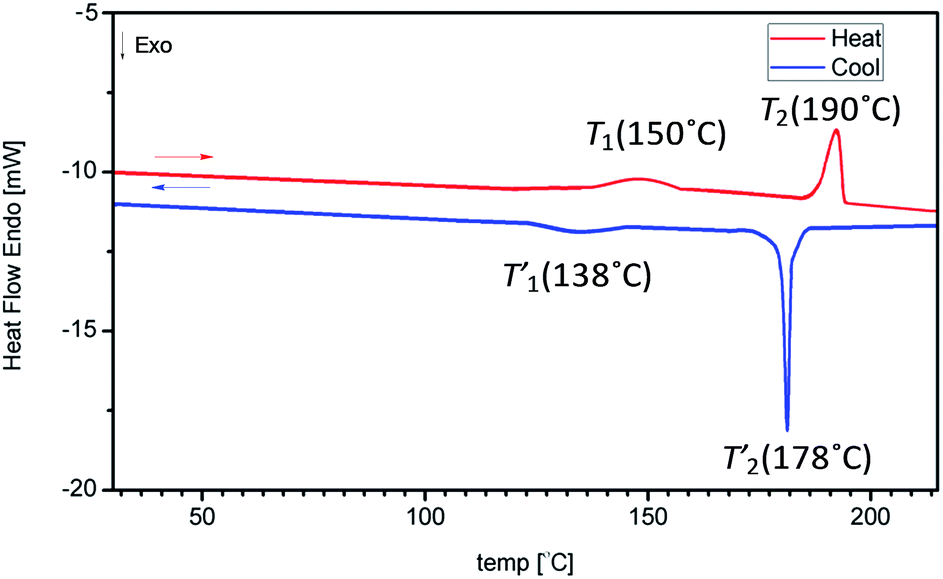 | ||
| Fig. 9 DSC thermogram of 4 for the second heating/freezing cycle. | ||
Besides the major phase transition (T2 = 190 °C) that is assigned for melting, an additional transition at 150 °C is observed. During this step the sample did not change the state of matter. Such a phenomenon is also observed during freezing and does not disappear over successive heating/freezing cycles. Similar occurrence was also noted by Heeley et al.27 during studies of T8-R (R = C14, C16, C18) SQs possessing long alkyl chains. In this case the above-mentioned phase transition has been classified as arising from chain rotation or as a minor structural reordering of the ends of the alkyl chain arms. Alkyl side groups during rotation cause minor reordering of the crystal lattice. Such an occurrence is not observed in the case of T10 compounds, probably because they possess too low crystallinity to detect such a sensitive transition by using DSC. For compounds which possess alkyl chains longer than C7 (alkanes), such a phenomenon is called a solid–solid phase transition.28
Transparency and hydrophobic properties
Due to the low molecular symmetry of the resulting 4 and 5 containing long alkyl amido-functionalized side-chain groups, their structure led to the formation of optically transparent films. Hence, compounds 4 and 5 were used as coatings on a glass substrate. As shown in Fig. 10a, organic–inorganic hybrids are in fact transparent and their transparency in the visible light region was confirmed by UV-vis spectroscopy. It is worth noting that the low crystallinity of 4 and 5, confirmed by powder XRD and TEM analyses (see ESI Fig. S39, S48, and S60†), strongly affects such transparency and might be regarded as an undisputed advantage of these materials. A similar phenomenon was observed by Kaneko et al. in the case of low-crystalline POSS containing two types of alkylammonium substituents.29 The low state of crystallinity derives from the fact that the compounds possess long alkyl chains. | ||
| Fig. 10 (a) UV-vis spectra of 4 (black line) and 5 (red line). (b) 4 and (c) 5 coated on the glass plate (the amount of each product on the surface is ca. 3.0 mg cm−2). The insets show photographs exhibiting the interaction of water with a surface coated by 4 (b) and 5 (c) films. | ||
Moreover, due to the lamellar-like nature of long side-chains, the created surfaces exhibited hydrophobic properties. The values of contact angles range from 104° to 110° for 4 and 5, respectively (Fig. 10b and c insets). Compounds 1–3 are soluble in water and contact angles could not be measured. Hydrophilic glass plates before coatings were also wetted with water and formation of drops was not detected due to a very low contact angle.
Additionally, the difference between crystallization and melting temperatures is ca. 10 °C, which clearly corresponds to the formation of well-organized structures (so-called self-assembled long-range ordering) on the glass surface.30 Such a difference in temperatures (ΔT = T0m − T0f) is regarded as the degree of supercooling and is required to initiate the crystallization process in the lamellar microdomains.30c Based on our best knowledge, the only example of alkylated cage silsesquioxane forming with a hierarchical lamellar nanostructure was reported by Hayakawa et al.31
Conclusions
In conclusion, we reported the synthesis and characterization of the novel ionic crystalline deca(3-aminopropyl)silsesquioxane trifluoromethanesulfonate 3, low crystalline non-fluorinated amido-functionalized octa(3-decanamidopropyl) 4 and deca(3-decanamidopropyl)silsesquioxane 5. Compounds 4 and 5 represent a hitherto unstudied group of SQs with long alkyl side-chains that were successfully isolated. Here, we demonstrated that 13C NMR experiments are helpful in interpreting hexa- and heptahedral SQs and constitute a much more useful tool compared to silicon 29Si NMR. Also, DOSY studies delivered essential data which allowed differentiation of compounds with different mass and symmetry. Moreover, in this study we demonstrated useful synthetic method that allowed us to obtain numerous intermediate compounds that can be trapped during hexahedral T8 → heptahedral T10 cage-rearrangement. All isolated species were characterized by 29Si NMR spectroscopy and high-resolution mass spectrometry.We have presented thermal analysis of 4 and 5 and revealed their transparency and hydrophobic properties that can be useful in various industrial applications. Octa 4 and decamer 5 silsesquioxanes decompose at 471 and 451 °C, respectively, which emphasizes their higher thermal stability compared to non-functionalized derivatives and organic species. Moreover, molecular structures of 4 and 5 allow the formation of materials that are optically transparent in the visible light region and hydrophobic films. It is possible because long alkyl amido-functionalized side chains attached to the siloxane core probably form the lamellar-like surfaces. The values of contact angles range from 104 to 110° for 4 and 5, respectively. It should be noted that due to the difference between crystallization and melting points, which equals ca. 10 °C, the formation of well-organized structures is observed, which is not a common phenomenon in this class of compounds.
Acknowledgements
This work was supported by the National Research Centre, Poland (Grant No. 2013/09/D/ST8/03995).Notes and references
- V. A. Ganesh, H. K. Raut, A. S. Nair and S. Ramakrishna, J. Mater. Chem., 2011, 21, 16304 RSC.
- P. Ragesh, V. A. Ganesh, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 14773 CAS.
- V. A. Ganesh, S. S. Dinachali, A. S. Nair and S. Ramakrishna, ACS Appl. Mater. Interfaces, 2013, 5, 1527 CAS.
- D. Xiong, G. Liu and E. Scott, J. Polym., 2013, 54, 3008 CrossRef CAS PubMed.
- C. Y. Mou, W. L. Yuan and C. H. Shih, Thin Solid Films, 2013, 537, 202 CrossRef CAS PubMed.
- G. R. Choi, J. Park, J. W. Ha, W. D. Kim and H. Lim, Macromol. Mater. Eng., 2010, 295, 995 CrossRef CAS PubMed.
- Some examples: (a) R. H. Baney, M. Itoh, A. Sakakibara and T. Suzuki, Chem. Rev., 1995, 95, 1409 CrossRef CAS; (b) D. A. Loy and K. J. Shea, Chem. Rev., 1995, 95, 1431 CrossRef CAS; (c) R. Duchateau, Chem. Rev., 2002, 102, 3525 CrossRef CAS PubMed; (d) R. Y. Kannan, H. J. Salacinski, P. E. Butler and A. M. Seifalian, Acc. Chem. Res., 2005, 38, 879 CrossRef CAS PubMed; (e) D. B. Cordes, P. D. Lickiss and R. Franck, Chem. Rev., 2010, 10, 2081 CrossRef PubMed , and references therein.
- (a) I. Imae and Y. Kawakami, J. Mater. Chem., 2005, 15, 4581 RSC; (b) M. F. Roll, M. Z. Asuncion, J. Kampf and R. M. Laine, ACS Nano, 2008, 2, 320 CrossRef CAS PubMed; (c) S. Sulaiman, A. Bhaskar, J. Zhang, R. Guda, T. Goodson III and R. M. Laine, Chem. Mater., 2008, 20, 5563 CrossRef CAS.
- Some examples: (a) R. M. Laine and M. F. Roll, Macromolecules, 2011, 44, 1073 CrossRef CAS; (b) K. L. Chan, P. Sonar and A. J. Sellinger, J. Mater. Chem., 2009, 19, 1 RSC.
- M. Z. Asuncion and R. M. Laine, J. Am. Chem. Soc., 2010, 132, 3723 CrossRef CAS PubMed.
- Some examples: (a) Y. Kawakami, K. Yamaguchi, T. Yokozawa, T. Serizawa, M. Hasegawa and Y. Kabe, Chem. Lett., 2007, 36, 792 CrossRef CAS; (b) A. R. Bassindale, Z. Liu, I. A. Mackinnon, P. G. Taylor, Y. Yang, M. E. Light, P. N. Horton and M. B. Hursthouse, Dalton Trans., 2003, 2945 RSC; (c) V. Ervithayasuporn, X. Wang and Y. Kawakami, Chem. Commun., 2009, 5130 RSC.
- T. Jaroentomeechai, P. Yingsukkamol, C. Phurat, E. Somsook, T. Osotchan and V. Ervithayasuporn, Inorg. Chem., 2012, 51, 12266 CrossRef CAS PubMed.
- J. Jung, H. J. C. Furgal, T. Goodson III, T. Mizumo, M. Schwartz, K. Chou, J.-F. Vonet and R. M. Laine, Chem. Mater., 2012, 24, 1883 CrossRef.
- M. Ronchi, S. Sulaiman, N. R. Boston and R. M. Laine, Appl. Organomet. Chem., 2010, 24, 551 CrossRef CAS PubMed.
- A. Miyazato, C. Pakjamsai and Y. Kawakami, Dalton Trans., 2010, 3239 RSC.
- J. C. Furgal, J. H. Jung, T. Goodson III and R. M. Laine, J. Am. Chem. Soc., 2013, 135, 12259 CrossRef CAS PubMed.
- V. Ervithayasuporn and S. Chimjarn, Inorg. Chem., 2013, 52, 13108 CrossRef CAS PubMed.
- M. Janeta, Ł. John, J. Ejfler and S. Szafert, Chem.–Eur. J., 2014, 20, 15966 CrossRef CAS PubMed.
- (a) F. J. Feher and K. D. Wyndham, Chem. Commun., 1998, 323 RSC; (b) F. J. Feher, K. D. Wyndham, D. Soulivong and F. Nguyen, J. Chem. Soc., Dalton Trans., 1999, 1491 RSC.
- M.-C. Gravel, C. Zhang, M. Dinderman and R. M. Laine, Appl. Organomet. Chem., 1999, 13, 329 CrossRef CAS.
- R. Weidner, N. Zeller, B. Deubzer and V. Frey, US Pat., 5,047,492, 1991.
- Y. Kaneko, M. Shoiriki and T. Mizumo, J. Mater. Chem., 2012, 22, 14475 RSC.
- S. Chimjarn, R. Kunthom, P. Chancharone, R. Sodkhomkhum, P. Sangtrirutnugul and V. Ervithayasuporn, Dalton Trans., 2015, 916 RSC.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081 CrossRef CAS PubMed.
- F. J. Feher, D. Soulivong and A. G. Eklund, Chem. Commun., 1998, 399 RSC.
- (a) G. Cheng, T. Hasell, A. Trewin, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 124, 12899 CrossRef PubMed; (b) H. Wang, Y. Xue, J. Ding, L. Feng, X. Wang and T. Lin, Angew. Chem., Int. Ed., 2011, 123, 11635 CrossRef PubMed; (c) K. Golovin, D. H. Lee, J. M. Mabry and A. Tuteja, Angew. Chem., Int. Ed., 2013, 125, 13245 CrossRef PubMed.
- E. L. Heeley, D. J. Hughes, Y. E. Aziz, P. G. Taylor and A. R. Bassindale, Macromolecules, 2013, 46, 4944 CrossRef CAS.
- R. Boese, H.-C. Weiss and D. Bläser, Angew. Chem., Int. Ed., 1999, 38, 988 CrossRef CAS.
- T. Tokunaga, M. Shoiriki, T. Mizumo and Y. Kaneko, J. Mater. Chem. C, 2014, 2, 2496 RSC.
- (a) H.-L. Chen, S.-C. Hsiao, T.-L. Lin, K. Yamauchi, H. Hasegawa and T. Hashimoto, Macromolecules, 2001, 34, 671 CrossRef CAS; (b) H.-L. Chen, J.-C. Wu, T.-L. Lin and J. S. Lin, Macromolecules, 2001, 34, 6936 CrossRef CAS; (c) J.-G. Li, C.-Y. Chung and S.-W. Kuo, J. Mater. Chem., 2012, 22, 18583 RSC.
- L. Wang, Y. Ishida, R. Maeda, M. Tokita, S. Horiuchi and T. Hayakawa, Langmuir, 2014, 30, 9797 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Multinuclear NMR (1H, 13C, 29Si), DOSY NMR, COSY NMR, FTIR spectra of 1–6, TG-DTA, DSC, HR-MS, powder XRD and HR-TEM-EDS. See DOI: 10.1039/c5ra10136k |
| This journal is © The Royal Society of Chemistry 2015 |