
An overview of Zn-catalyzed enantioselective aldol type C–C bond formation
Amrutha P. Thankachan
,
S. Asha
,
K. S. Sindhu
and
Gopinathan Anilkumar
*
School of Chemical Sciences, Mahatma Gandhi University, Priyadarsini Hills P. O., Kottayam, Kerala, India 686560. E-mail: anilgi1@yahoo.com; Fax: +91-481-2731036; Tel: +91-481-2731036
First published on 13th July 2015
Abstract
Enantioselective aldol reactions are powerful tools for the creation of carbon–carbon bonds in a stereoselective manner. Several synthetic strategies have been developed for asymmetric aldol reactions; but among these, Zinc-catalyzed protocols for enantioselective aldol reactions attained great significance due to its excellent functional group tolerance. Zinc catalysis has an undeniable significance over other catalytic systems due to its non-toxic, easily available, cheap and environmentally benign properties. This review mainly focuses on the stereoselective aspects of Zn-catalyzed aldol type reactions promoted by enolate chemistry.
 Amrutha P. Thankachan | Amrutha P. Thankachan was born in Kerala, India, in 1988. She obtained her B.Sc. degree from Mahatma Gandhi University (C M S College, Kottayam) in 2009 and her M.Sc. degree from School of Chemical Sciences, Mahatma Gandhi University in 2011. Currently she is doing her doctoral research under the guidance of Dr G. Anilkumar in School of Chemical Sciences, Mahatma Gandhi University. |
 S. Asha | S. Asha was born in Kerala, India, in 1988. She received her B.Sc. degree from Mahatma Gandhi University (Assumption College, Changanassery) in 2009 and her M.Sc. degree from School of Chemical Sciences, Mahatma Gandhi University in 2011. Currently, she is doing her doctoral research under the guidance of Dr G. Anilkumar in School of Chemical Sciences, Mahatma Gandhi University. |
 K. S. Sindhu | K.S Sindhu was born in Kerala, India, in 1983. She received her B.Sc. degree from Mahatma Gandhi University (St. Xaviers College, Aluva) in 2003 and her M.Sc. degree from School of Chemical Sciences, Mahatma Gandhi University in 2007. She earned National Eligibility test (NET) with a scholarship. Currently she is doing her doctoral research under the guidance of Dr G. Anilkumar in the School of Chemical Sciences, Mahatma Gandhi University. |
 Gopinathan Anilkumar | Gopinathan Anilkumar was born in Kerala, India and took his Ph.D in 1996 from Regional Research Laboratory, Trivandrum with Dr Vijay Nair. He did postdoctoral studies at University of Nijmegen, Netherlands (with Professor Binne Zwanenburg), Osaka University, Japan (with Professor Yasuyuki Kita), Temple University, USA (with Professor Franklin Davis) and Leibniz-institüt für Katalyse, Rostock, Germany (with Professor Matthias Beller). He was a senior scientist at AstraZeneca (India). Currently he is Associate Professor in Organic Chemistry at the School of Chemical Sciences, Mahatma Gandhi University in Kerala, India. His research interests are in the areas of organic synthesis, medicinal chemistry and catalysis, particularly on ruthenium, iron, zinc and copper catalyzed reactions. |
1. Introduction
Zinc is the 24th most abundant element found in the Earth's crust. The pure zinc metal was discovered by Andreas S Marggraf in 1746. The material zinc is mainly used for corrosion-resistance, biological and chemical applications. Among the different bond-forming reactions, the C–C bond formation is the most prominent one in organic synthesis. Some examples of C–C bond formation are the aldol reaction, Mannich reaction, Diels–Alder reaction and transition metal-catalyzed coupling reactions. The aldol reaction is one of the most effective and accepted methods for the formation of C–C bonds.1 Although the classical aldol reaction is atom-efficient, it has some drawbacks.2 Simple aldol reaction suffers from issues related to selectivity especially with regard to chemo- and regioselectivity. Another challenge is the implementation of this reaction in an asymmetric fashion. However, excellent accomplishments have been made in this area. Like other asymmetric reactions, this reaction also uses chiral auxiliaries, catalysts and ligands as stereo-controllers. Among these, chiral transformation by catalytic use of chiral-controllers is the most elegant one. The catalytic asymmetric reactions are achieved by the use of either biochemical catalyst or chemical catalyst.3 The chemical catalyst used for this purpose is usually a transition metal catalyst or a non-metallic simple organic molecule (organo catalyst). In transition metal-catalyzed asymmetric reactions, the pivotal role is played by the metal which induces chirality and improves reactivity due to the presence of chiral ligands.Due to the high abundance, non-toxic, cheap and environmentally benign nature of zinc salts, they have been used as catalyst in aldol reactions.4 Zinc is capable of forming bonds with greater covalency and it can also form stable complexes with N- and S-donors. Many molecules with asymmetric centers have excellent biological properties and consequently stereoselective aldol reactions rapidly attain great importance in synthetic organic chemistry. Of the numerous drugs available nowadays, many have at least one asymmetric center.5 In short this newly introduced method of asymmetric aldol reaction allows straight forward enantioselective entry into a number of natural products. Although a number of reviews dealing with enantioselective versions of aldol type reactions are available,6–9 the one dealing with enantioselective zinc-catalyzed aldol type reaction is rare.4 Very recently, the applications of chiral zinc catalyst in asymmetric synthesis have been reported and the present review exclusively covers all the aspects of zinc-catalyzed enantioselective aldol reactions in details.10 This review focuses on Zn-catalyzed enantioselective aldol type reaction taking place via enolate chemistry and covers literature from 1980–2015.
2. Zn-catalyzed C–C bond formation reactions
Since C–C bond is the most fundamental bond in organic chemistry and is present in the basic skeleton of almost all organic molecules, methods leading to the formation of carbon–carbon bond are of great significance. Various methodologies are available to realize the C–C bond formations; some examples are the Diels–Alder cycloaddition,11 aldol reaction,12 transition metal-catalyzed coupling reactions etc.13,14 Among the different transition metal-catalyzed coupling reactions, the most extensively studied one is the palladium-catalyzed coupling reaction.15 Since palladium is toxic and expensive, other transition metals such as Cu,16 Ni,17 Co18 and Fe19 were introduced for coupling reactions. Later Zn has been identified as a suitable catalyst for C–C coupling reactions. These zinc-catalyzed reactions can be classified into non-stereoselective and stereoselective reactions.2.1 Zn-catalyzed non-stereoselective reactions
Zinc-catalysis is now one of the most promising areas of transition metal-catalyzed coupling reactions. This is because zinc salts are cheap, easily available, non-toxic and have eco-friendly behaviour. The use of organo-zinc reagents in organic synthesis has been known for long time. Some examples of reactions involving organo-zinc reagents are the Reformatsky reaction,20–22 Negishi coupling,23 Fukuyama reaction24 and so on. There are a large number of examples for non-stereoselective zinc-catalyzed reactions.25 Among the different organo-zinc reagents, the most popular one is the diethyl zinc which is one of the oldest reagents in synthetic organic chemistry.262.2 Zn-catalyzed stereoselective reactions
Asymmetric catalysis has achieved great attention for the past few years and contributed widely in the development of synthetic organic chemistry. The stereochemically controlled aldol reaction has attracted considerable interest over the last few years, as it is one of the most fundamental reactions in organic chemistry for carbon–carbon bond formation. The stereoselectivity in aldol reactions is attained by bio-chemical catalysts, transition metal catalysts or non-metallic organic molecules.27 A detailed account of zinc-catalyzed enantioselective aldol type reactions, their reaction conditions, green chemistry approach, mechanistic study and applications are discussed here.2.3 Zn-catalyzed stereoselective aldol reactions
Aldol reaction is one of the well-known reactions for C–C bond formation.1 The first report on zinc-catalyzed aldol-reaction was revealed by Watanabe and coworkers in 1980 while examining the catalytic ability of first row transition metal(II)-complexes.28 The results showed that Zn(II), Cu(II), Ni(II) and Co(II) acetates in presence of 2,2′-bipyridine ligands afforded the aldol products in moderate yields. The Zn-catalyst system was found to be compatible with aqueous solvent. Inspired by the result, the same group also conducted aldol condensation of p-nitrobenzaldehyde with acetone using some first and second row transition metal(II) complexes of α-amino acid esters.29 The Zn(II)-complexes of tyrosine ethyl ester showed maximum catalytic activity with moderate yield of the product; but no asymmetric induction was obtained.The first enantioselective aldol reaction was reported by Trost et al. in 2000.30 They developed a novel dinuclear zinc-complex derived from prolinol substituted phenol (Fig. 1) as an effective catalyst for aldol reaction of aryl methyl ketones and aldehydes (Scheme 1). Moderate yield of the asymmetric aldol product with excellent enantioselectivity was obtained in THF in the presence of the zincaphilic additive triphenylphosphine sulfide and 4 Å molecular sieves (the additives compatible with zinc catalytic system are known as zincaphilic additives).
 | ||
| Fig. 1 Bis(prolinol)phenol ligand and dinuclear Zn catalyst. | ||
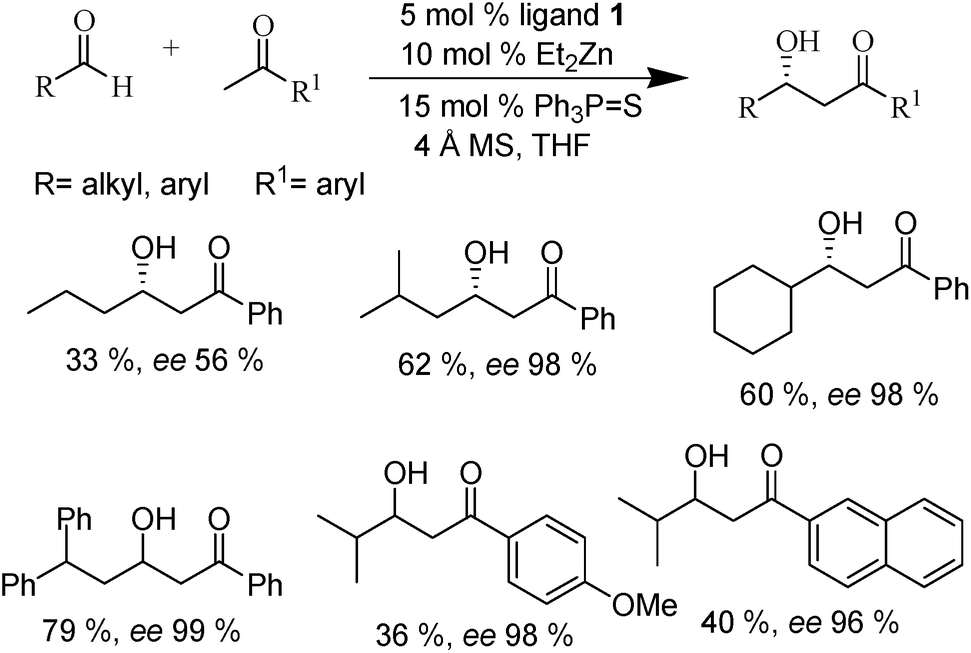 | ||
| Scheme 1 Enantioselective aldol reactions of aryl methyl ketones with aldehydes. | ||
The same catalytic system was also used for aldol reactions of α-hydroxyketones and aldehydes.31 Increasing the size of α-substituent of aldehydes increased the diastereoselectivity of the aldol product (Scheme 2). On replacing the phenyl substituent of hydroxyacetophenone with furan showed excellent improvement in enantiomeric excess (ee) but no influence on the diastereomeric ratio (dr). As expected, temperature also has considerable effect on ee. On lowering the reaction temperature, the ee increased without affecting the value of dr. Later Trost used the novel dinuclear zinc catalyst 2 for the stereoselective synthesis of (+)-boronolide.32
 | ||
| Scheme 2 Dinuclear zinc-catalyzed asymmetric aldol reactions of α-hydroxyketones. | ||
Later Trost applied similar catalytic system for the asymmetric aldol reaction of acetone with various aldehydes (Scheme 3).33 The results showed that a modified second generation ligand (3) was found to be superior to the previously used ligand. Analysis of the substituent effect of the aldehydes showed that α-branched aldehydes afforded higher yield and selectivity compared to the α-unbranched aldehydes.
 | ||
| Scheme 3 Zinc-catalyzed aldol reaction of acetone with various aldehydes. | ||
One of the challenges of this dinuclear catalytic system was the asymmetric aldol reaction of ynones. Later this was also achieved by Trost by performing enantioselective aldol reaction between methyl ynones and ketals of pyruvaldehyde.34 In the optimized reaction condition, THF was used as the solvent at 0 °C in the presence of 4 Å molecular sieves. Thus α,α-diethoxy aldehydes having allyl, methallyl and protected hydroxymethyl groups were reacted with methyl ynones affording the aldol product in moderate yield and enantioselectivity (Scheme 4).
 | ||
| Scheme 4 Enantioselective aldol reaction of methyl ynones. | ||
Using the same catalytic system Trost's group also reported an atom-economical route for the synthesis of β-hydroxy enones.35 Methylvinylketone reacted with differently substituted aldehydes in the presence of 10 mol% of the catalyst 2 and 4 Å MS in toluene or THF at low temperature affording the β-hydroxy enones in moderate yield and good enantioselectivity (Scheme 5). The reaction was found to be faster in THF, but toluene showed greater control over the elimination byproduct. Aldehydes with aliphatic and protected α- or β-hydroxy substituents gave stereoselective aldols with excellent selectivity. The hydroxyenones obtained can be easily transformed into useful molecular frameworks.
 | ||
| Scheme 5 Dinuclear zinc-catalyzed enantioselective aldol reaction of methylvinyl ketone. | ||
At the same time, Shibasaki's group also developed other dinuclear catalysts derived from BINOL for enantioselective aldol reactions of α-hydroxyacetophenone.36 The results showed that lanthanide dinuclear catalysts afforded exclusively the anti-α,β-dihydroxy ketones with high ee while zinc dinuclear catalysts gave the corresponding syn-α,β-dihydroxy ketones with excellent ee (Scheme 6). The nature of the substituents on the aldehyde did not show significant effect on the yield and ee.
 | ||
| Scheme 6 Asymmetric aldol reactions of α-hydroxyacetophenone catalyzed by Zn–Zn-linked BINOL complex. | ||
A comparative study of the catalytic ability of different Et2Zn/(S,S)-linked BINOL systems (Fig. 2) were also reported by the same group.37 The results showed good yield and stereoselectivity for simple Et2Zn/(S,S)-linked BINOL system in comparison to Et2Zn/(S,S)–sulfur-linked and Et2Zn/(S,S)–carbon-linked BINOL systems. The sulfur and carbon linked BINOL systems exhibited very good yield but the selectivity was poor (Table 1). On the basis of catalyst loading, 5a provides the most efficient small molecular catalyst for direct enantioselective aldol reaction.38 The syn-aldol adduct produced via this method was later transformed into an ester and an amide by regioselective rearrangement.
 | ||
| Fig. 2 Et2Zn/(S,S)-linked BINOL systems. | ||

Darbre's group introduced new ligand systems with N5 and N3O pattern for Zn-catalyzed aldol reaction of 2-hydroxyacetophenone and benzaldehyde39 (Fig. 3). The ligands 2,6-bis{[(pyrid-2-ylmethyl)amino]methyl}pyridine, N5 (6) and (6-{[(pyrid-2-ylmethyl)amino]methyl}pyrid-2-yl)methanol, N3O (7) afforded aldol products with yields ranging from 17–62%. Among the different zinc salts investigated, zinc acetate and diethyl zinc gave better yield. The selectivity of the aldol adduct was determined by 1H NMR analysis.
 | ||
| Fig. 3 Structure of N5 and N3O ligands. | ||
Darbre et al. also reported that Zn–proline complex acts as an efficient catalyst for the direct aldol reaction of a wide variety of aryl and heteroaryl carbaldehydes and acetone.40,41 The reactions were performed at room temperature in acetone–H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) solvent system (Scheme 7). The catalytic abilities of other amino acid–zinc complexes were also investigated which revealed that Zn–lysine and Zn–arginine complexes could also act as catalysts for asymmetric aldol reactions. It was observed that aryl aldehydes containing electron withdrawing or electron releasing substituents gave good yields and moderate ee of the aldol product. The ee values of ortho substituted aryl carbaldehydes were lower compared to the corresponding para-isomers. The same catalytic system was also applied for the aldol reactions of heterocyclic aldehydes, hydroxy and dihydroxyacetones. Later the Zn–proline catalytic system was used for the enantioselective synthesis of natural D-tetrose, pentose, hexose and small sugars.42,43
:2) solvent system (Scheme 7). The catalytic abilities of other amino acid–zinc complexes were also investigated which revealed that Zn–lysine and Zn–arginine complexes could also act as catalysts for asymmetric aldol reactions. It was observed that aryl aldehydes containing electron withdrawing or electron releasing substituents gave good yields and moderate ee of the aldol product. The ee values of ortho substituted aryl carbaldehydes were lower compared to the corresponding para-isomers. The same catalytic system was also applied for the aldol reactions of heterocyclic aldehydes, hydroxy and dihydroxyacetones. Later the Zn–proline catalytic system was used for the enantioselective synthesis of natural D-tetrose, pentose, hexose and small sugars.42,43
 | ||
| Scheme 7 Zn–proline catalyzed aldol reactions of acetone. | ||
The first zinc-catalyzed enantioselective aldol reaction between aryl aldehydes and aryl ketones was reported by Da and co-workers.44 In the optimized reaction condition, 20 mol% of ligand and 40 mol% of Et2Zn were used in the presence of 4 Å MS in DMF at 0 °C (Scheme 8). Here, triethylamine is also used as a zincaphilic additive. meta-Substituted aldehydes showed higher selectivity than ortho- and para-substituted aldehydes. Strong electron withdrawing and electron donating substituents in the aryl ring reduced the selectivity. The same catalytic system was also used for the asymmetric aldol reaction of heterocyclic aldehydes.
 | ||
| Scheme 8 Chiral-BINOL derived zinc-catalyzed stereoselective aldol reaction of aryl aldehydes and aryl ketones. | ||
In biological systems, naturally occurring enzyme aldolase acts as a catalyst for the reversible and stereospecific aldol reactions.45 The class-I aldolases catalyse the aldol reaction via enamine intermediate while the class-II aldolases catalyse via the formation of Zn2+ enolates. Inspired by the class-II aldolase chemistry, Aoki et al. tried catalytic enantioselective aldol reactions of Zn2+ complexes of L-prolyl-pendant[15]ane N5 (9), L-prolyl-pendant[12]ane N4 (10) and L-valyl-pendant[12]ane N4 (11) (Fig. 4).46
 | ||
| Fig. 4 Structure of L-prolyl-pendant[15]ane, L-prolyl-pendant[12]ane and L-valyl-pendant[12]ane ligands. | ||
Among these three catalytic systems, 9 and 10 gave better yield and enantioselectivity. Acetone–water (4:1) solvent system at 37 °C was found to be the best condition for the reaction and was applied in the aldol reaction of acetone, hydroxy acetone and dihydroxy acetone with aromatic aldehydes (Scheme 9).
 | ||
| Scheme 9 Zinc-catalyzed stereoselective aldol reaction of acetone and aryl aldehydes. | ||
Zn(pybox)-catalyzed asymmetric Mukaiyama aldol reaction between aldehydes and silyl enol ethers has been reported.47 Under the optimized reaction condition (20 mol% catalyst with THF–H2O (9:1) solvent system at 0 °C), aromatic, α,β-unsaturated and aliphatic aldehydes afforded the corresponding β-hydroxyketones in good yield and stereoselectivity (Scheme 10). The catalyst was also recovered after the reaction.
 | ||
| Scheme 10 Zn-(pybox)-catalyzed asymmetric Mukaiyama aldol reaction. | ||
Zn-supported enantioselective aldol reaction in aqueous media has been reported using a series of prolinamide-based chiral ligands (Scheme 11).48 Among the different prolinamide ligands, 14 gave excellent yield and enantioselectivity under optimized reaction conditions. Aromatic aldehydes with electron withdrawing substituents afforded β-hydroxy aldol products with high yield and enantioselectivity. Heterocyclic aldehydes also furnished the aldol adducts with comparable yield and selectivity. The same catalytic system was also found to be suitable for acyclic ketones. Scale up of the reaction for large scale production retained the enantioselectivity.
 | ||
| Scheme 11 Zn–prolinamide catalysed enantioselective aldol reactions in aqueous media. | ||
Later organic solvent-free conditions were introduced in the field of zinc-catalyzed enantioselective aldol reactions.49 A combination of zinc triflate and prolinamide was introduced as a catalytic system for stereoselective aldol reactions (Scheme 12).50 The reaction was carried out using chiral derivative of 4-pyridyl substituted hydroxy prolinamide as the ligand in a mixed solvent system of cyclohexanone and water (1:1). The catalyst loading studies revealed that 20 mol% of Zn(OTf)2 afforded the highest yield and enantioselectivity. The investigation on the generality of the catalytic system showed that only aromatic aldehydes with electron withdrawing substituents afforded better yield and stereoselectivity. A modified version of the catalyst system with C1-symmetric chiral ligand was later introduced.51
 | ||
| Scheme 12 Zn–prolinamide catalysed enantioselective aldol reactions of cyclohexanone and aromatic aldehydes in aqueous media. | ||
2.4 Zn-catalyzed stereoselective Henry reaction
The Aldol type condensation reaction between nitroalkanes and carbonyl compounds affording β-nitroalcohols is known as the Henry reaction or nitroaldol reaction.52 The product of Henry reaction, the β-nitroalcohol, is an efficient precursor for β-hydroxy amines and β-hydroxy carboxylic acids. Even though the Henry reaction was reported in 19th century, the Zn-catalyzed version was reported only in 2002 by Trost's group53 which afforded the product in the range 56–90% yield and up to 93% ee.54 The dinuclear zinc-catalyst 2 (Fig. 1) is one of the reliable catalysts for asymmetric Henry reaction and efficiently catalyses the coupling between nitromethane and differently substituted aldehydes in THF at low temperature (Scheme 13). As expected, lower temperature enhanced the ee of the product. The addition of molecular sieves also improved the yield and ee of the product. α-Substituted aldehydes gave high yield and ee. This dinuclear catalyst was also found to be effective for the Henry reaction of heterocyclic aldehydes. | ||
| Scheme 13 Dinuclear Zn-catalyzed Henry reactions of nitromethane. | ||
Trost also made a comparative study on the catalytic efficiency of variously substituted ligands of the dinuclear Zn complex for Henry reaction (Scheme 14).55 The results revealed that varying the substituents on the phenol ring did not show any influence on the yield and enantioselectivity except in the case of p-methoxy and p-fluoro groups; the p-methoxy substituent on the phenol ring of the ligand decreased the yield and enantioselectivity while fluorine at the p-position caused a slight increase in ee. The naphthyl and biphenyl substituted dinuclear Zn complexes gave a slightly better enantioselectivity. The modified ligands were applied in the synthesis of the β-receptor agonists (−)-denopamine and (−)-arbutamine.
 | ||
| Scheme 14 Dinuclear Zn-catalyzed Henry reactions of nitromethane. | ||
Trost et al. also reported that the dinuclear zinc-catalyst effectively catalyzes aza–Henry reaction of nitroalkanes with differently protected imines.56 The aza–Henry adducts formed were later successfully transformed into 1,2-diamines or α-amino acids. Under optimized conditions, the reaction showed great sensitivity to electronic effects (Scheme 15). Decrease in enantioselectivity was observed when electron rich p-anisaldehyde derivative of imine was used. Heteroaromatic and α,β-unsaturated imines underwent the reaction smoothly. Boc-, Moc- and Cbz-protected imines and a variety of nitroalkanes were found to be good substrates for the reaction.
 | ||
| Scheme 15 Zn-catalyzed asymmetric Aza–Henry reaction. | ||
A new class of chiral macrocyclic ligands containing amino and thiophene moieties were introduced for Zn-catalyzed enantioselective Henry reaction (Fig. 5).57 Among the different ligands studied, the trimeric variety of 19 provided better yield and selectivity.
 | ||
| Fig. 5 Structure of chiral macrocyclic ligands. | ||
Palomo and co-workers also introduced a successful Zn-catalytic system for the enantioselective Henry reaction.58 In the optimum reaction condition, a combination of Zn(OTf)2, i-Pr2EtN and (+)-N-methylephedrine was used in the ratio 30:30:45. Both aromatic and aliphatic aldehydes were tolerated in the reaction with equal chemical efficiency; but the enantioselectivity was less for aromatic aldehydes compared to aliphatic aldehydes. The same catalytic system was also applicable for enantioselective aza–Henry reactions.59
2.5 Zn-catalyzed stereoselective Mannich reactions
Mannich reaction (imine Aldol reaction) is one of the important C–C bond forming reactions in organic chemistry leading to the formation of β-amino carbonyl compounds.60 The asymmetric Mannich reaction provides an innovative route for the generation of α- and β-amino acid derivatives, β-lactams and γ-amino alcohols. It is also used an excellent method for the preparation of nitrogen containing biologically active compounds. The first Zn-catalyzed Mannich type reaction in aqueous medium was reported by Kobayashi et al. in 2002.61 They reported that Mannich type reactions of hydrazo esters with silyl enol ethers resulted in the formation of the respective acylhydrazones (Scheme 16). Among the different Zn-salts studied, ZnF2 gave better enantioselectivity albeit in low yields. In the presence of 1 mol% of trifluoromethanesulfonic acid (TfOH), the ZnF2 afforded good yield and high enantioselectivity. Both aromatic and aliphatic silyl enol ethers gave the Mannich products in 30–91% yield and 88–91% enantioselectivity. The sphingomyelin inhibitor HPA-12 was successfully synthesized utilizing this methodology. | ||
| Scheme 16 Zn-catalyzed asymmetric Mannich reaction. | ||
Trost et al. successfully used the dinuclear zinc catalyst 2 and 21 for asymmetric Mannich reaction.62 Remarkable influence of electronic and steric effects of ligand substituents was observed for this reaction (Scheme 17). Compared to phenyl substituent on the dinuclear catalyst, the biphenyl substituent gave better yield and selectivity. The electron-rich hydroxy ketones required higher catalyst loading. The electron withdrawing substituents in the imine increased the yield and selectivity. The catalytic system was also applied to heteroaromatic compounds.
 | ||
| Scheme 17 Dinuclear zinc-catalyzed asymmetric Mannich reactions. | ||
Shibasaki's dinuclear Zn–BINOL system, 5a (Fig. 2) also catalyzed asymmetric Mannich reaction; but the diastereoselectivity was found to be opposite to that observed for Aldol reaction.63 Due to the synchronized formation of two adjacent stereocenters, asymmetric Mannich reaction of α-alkoxy enolates has great importance in organic synthesis. Mannich reaction of 2-hydroxy-2′-methoxyacetophenone with N-diphenylphosphenoyl (Dpp) imines exclusively gave the anti-alcohols with comparable yield and enantiomeric excess (Table 2). In the optimized reaction condition (1 mol% of ligand, 4 mol% of Et2Zn and 3 Å MS were used in THF at −20 °C), imines derived from α-nonenolizable aldehydes and aromatic ketones with ortho-substituent resulted exclusively the anti-adduct with high ee. The imines derived from α,β-unsaturated aldehydes gave adducts with low dr.
|
|||
|---|---|---|---|
| R | Yield (%) | dr (anti/syn) | ee (%) (anti) |
| 4-Me–C6H4– | 98 | 96/4 | 98 |
| 2-Me–C6H4– | 99 | >98/2 | 99 |
| 2-Naphthyl | 95 | 94/6 | 99 |
| E-(Cinnamyl) | 98 | 76/24 | >99.5 |
| Cyclopropyl | 98 | 80/20 | 99 |

The same catalytic system was also used for the synthesis of syn-β-amino alcohols through stereoselective Mannich reaction by protecting the amine with Boc group.64 The optimized reaction condition for syn-selectivity was the same as that for anti-selectivity; but maximum diastereoselectivity was obtained at −40 °C (Table 3). The results revealed that imines derived from both aromatic and heteroaromatic moieties gave better yield and selectivity. But imines from 3-pyridyl species afforded products in low yield and moderate selectivity. The diastereoselectivity was less for imines from E-cinnamyl and 2-allylfuryl moieties. Later Trost et al. also reported similar results with their dinuclear Zn–prolinyl phenol complex 2.65
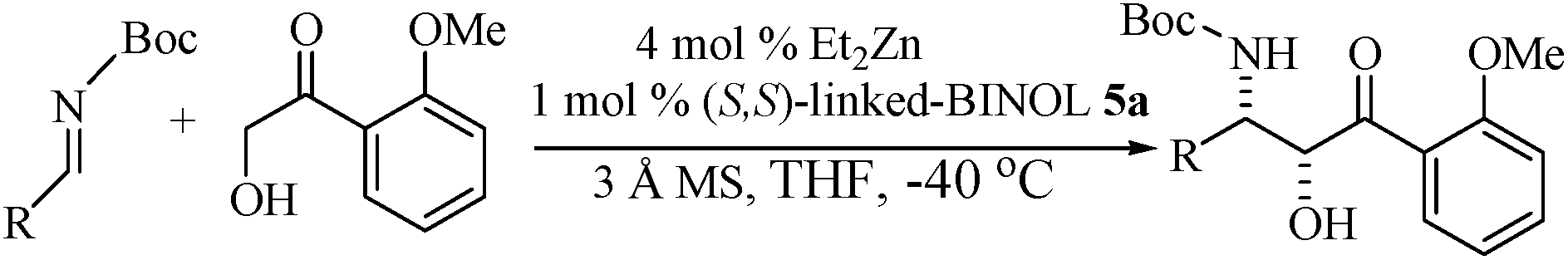
Zinc-catalyzed enantioselective Mannich-type-reaction of hydrazones with difluoroenoxysilanes was reported by Shi et al.66 The reaction was carried out in the presence of 10 mol% of Zn(NTf2)2, 20 mol% of imidazoline-anchored phosphine ligand in THF/MeOH(1:1) at 5 °C (Scheme 18). For further improvement in enantioselectivity the zincaphilic additive 4 Å MS was also used. This catalytic system was found to be superior to the previously reported Zn(OTf)2–oxazoline–phosphine-ligand for asymmetric difluorination of hydrazones with difluoroenoxysilanes.67 Incorporation of fluorine atoms into the target molecule resulted large enhancement in their physical, chemical and pharmacological activity.68 Here the electronic nature of the substituent did not have great influence on the yield and enantioselectivity of Mannich adduct. The present catalytic system is also applicable for difluorination of aliphatic hydrazones.
 | ||
| Scheme 18 Zn-catalyzed asymmetric Mannich-type reaction of hydrazones with difluoroenoxysilanes. | ||
2.6 Zn-catalyzed stereoselective Michael reactions
As in the case of Aldol and Mannich reactions, Michael reaction also provides an excellent route for C–C bond formation.69 In 2003 Shibasaki's group reported asymmetric Michael addition using Zn-catalysis.70 They used the first and second generation Et2Zn/(S,S)-linked-BINOL system as catalyst for the reaction (Table 4). The second generation catalyst was found to be better compared to the first generation in terms of yield. It also offered a better method for the formation of 1,5-dicarbonyl compounds.
Trost's dinuclear zinc–prolinylphenol complex also showed excellent catalytic activity in asymmetric Michael-type reactions.71 Reaction of 5-methyl-2-phenyloxazol-4(5H)-one with β-nitrostyrene in the presence of dinuclear zinc–prolinylphenol catalyst afforded the Michael product in excellent yield (Scheme 19). The results revealed that the nature of the aryl substituent on zinc–prolinylphenol complex affects the yield and stereoselectivity of the reaction. The electron withdrawing substituent on the aryl ring did not show considerable change in yield and selectivity. The ligand with 2-naphthyl substituent provided complete conversion with better dr and ee values. Substrates with m-substituent on the aryl group of oxazolone furnished the product with good dr and excellent ee values compared to the corresponding o- and p-substituted compounds. Aryl substituted nitro olefins showed better results compared to alkyl, alkynyl and heteronuclear substituted compounds. Electron donating substituents at the p-position of the aryl group in nitro olefin gave higher yield compared to electron-withdrawing groups at the same position.
 | ||
| Scheme 19 Asymmetric Michael-type reactions catalyzed by dinuclear zinc–prolinylphenol complex. | ||
3. Green chemistry approach in enantioselective aldol reactions
Presently, one of the key objectives for synthetic organic chemists is the development of eco-friendly protocols for carrying out chemical reactions with the production of least amount of waste. Green chemistry adheres to a group of regulations such as avoiding the use of toxic and volatile solvents, reducing the amount of catalyst and reagents needed, achieving atom-economy, least usage of energy at optimized reaction conditions and production of minimal quantity of chemical waste.72 These requirements for greenness impart more pressure for achieving bio-active substances in environmentally compatible solvents.73 From green chemistry point of view the better method of performing chemical reaction would be under solvent-less conditions. But solvents are crucial for mass and heat transfer occurring during the reaction. Great efforts have been devoted to discover sustainable reaction media and thus the use of aqueous solvents have drawn considerable interest recently.74 Being a sustainable solvent, water has many advantages over other solvents such as enhancement in reactivity and selectivity, easy work-up, catalyst recycling, protecting group-free synthesis and mild reaction conditions. A large number of reports are available on Zn-catalyzed aldol reactions in aqueous media.8 Mlynarski reported an efficient Zn(pybox)-complex for asymmetric Mukaiyama aldol reactions in aqueous media.47 Here the catalyst was recycled after the first cycle and the same was extracted into the water phase and later recovered by concentration. Other successful methods for enantioselective aldol reactions in aqueous medium are also available.49–51,74 Zn–prolinamide complexes were effectively used as catalyst for Zn-assisted asymmetric aldol reaction in aqueous medium and were found amenable to large scale preparation.484. Mechanistic studies
The development of zinc catalyst afforded an atom economical route for stereoselective aldol reaction. This helped to overcome the commonly encountered challenges related to stereoselective aldol reactions. The enzymes that catalyze aldol reactions in biological system are called aldolases.75 The aldolases catalyze the reaction either by type-I or type-II mechanism. In type-I aldolase the amino acid residue interacts with donor species to generate an enamine. The enamine later attacks the acceptor electrophile to yield an iminium adduct, which then undergoes hydrolysis affording the aldol adduct. But in type-II aldolase, the activation of donor species is by complexation with zinc (Scheme 20). This complexation makes the α-proton of carbonyl more acidic and subsequently generation of zinc enolate takes place. The attack of zinc enolate to the carbonyl group of acceptor electrophile which is already activated via hydrogen bonding results in the generation of zinc complex of aldol product. Finally the protonation and decomplexation give the respective aldol product. This natural process offers an insight into the mechanism of zinc-catalyzed stereoselective aldol reaction. | ||
| Scheme 20 Type II aldolase mechanism represented by fuculose-1-phosphate aldolase involving zinc enolate℗ = PO32− (reprinted with permission from Angew. Chem. Int. Ed., 2000, 39, 1352 C.-H. Wong et al.). | ||
In order to establish the stoichiometry of metal to ligand, Trost et al. conducted a detailed study on the mechanism of the Zn-catalyzed aldol type reaction.30 The structure of the catalyst was analyzed by measuring the amount of ethane gas liberated. The addition of 2 equivalents of diethylzinc per ligand generated the activated zinc catalyst along with 3 equivalents of ethane gas (Scheme 21). This observation indicated that the initial catalyst was a bimetallic moiety. Later the dinuclear catalyst reacted with another carbonyl species to evolve the fourth equivalent of ethane and commenced the catalytic cycle. The structure of the catalyst was confirmed by X-ray analysis.
 | ||
| Scheme 21 Trost's proposed catalytic cycle for asymmetric aldol reactions (reprinted with permission from J. Am. Chem. Soc, 2000, 122, 12003 Trost et al.). | ||
Shibasaki and co-workers performed a detailed mechanistic study on Et2Zn/(S,S)-linked-BINOL catalyzed aldol and Mannich reactions.37 Initially they postulated that a bimetallic monomer of Et2Zn/(S,S)-linked-BINOL was the catalytic species. But the X-ray crystallographic analysis and kinetic studies revealed the existence of trinuclear (Fig. 6) and heptanuclear zinc species. The presence of this multinuclear species in the reaction was also verified by 1H NMR and cold spray ionization-mass spectrometry (CSI-MS). It was finally concluded that zinc-alkoxide species play a central role in the enhancement of reaction rate and the expected rate determining step would be the product dissociative step to regenerate the Zn/linked BINOL/ketone oligomeric species. The proposed catalytic cycle for Et2Zn/(S,S)-linked-BINOL complex catalyzed aldol reaction is shown in Scheme 22.
 | ||
| Fig. 6 Structure of the trinuclear Zn complex Zn3(linked-BINOL)2(THF)3. | ||
 | ||
| Scheme 22 Proposed catalytic cycle for Et2Zn/(S,S)-linked-BINOL complex catalyzed direct aldol reaction (reprinted with permission from J. Am. Chem. Soc. 2004, 126, 8777 Shibazaki et al.). | ||
The oligomeric zinc-rich putative complex (I) is formed in the presence of preformed Zn3(linked-BINOL)2(THF)3 complex and ketone. This Zn–binaphthoxide complex is assumed to act as a BrØnsted base and deprotonate the α-proton of ketone resulting in the formation of II. At the same time aldehyde approaches from the Re-face of the enolate selectively activated by the zinc-Lewis acid center forming III, which immediately undergoes 1,2-addition to form IV. The protonation via phenolic proton of ligand followed by ligand exchange with ketone regenerates I. This would react smoothly with IV through transmetallation leading to the formation of the required syn-adduct. Similar catalytic cycles are postulated for both syn- and anti-stereoselective Mannich reaction catalyzed by the same Et2Zn/(S,S)-linked-BINOL complex.64
Darbre and co-workers proposed a catalytic cycle for Zn–proline catalyzed stereoselective aldol reaction in aqueous medium.41 Accordingly, the metal in the catalyst acts as a Lewis acid in water which imitates the naturally occurring class-II aldolases. It is observed that the zinc–proline complex is the catalytic species and not zinc or proline alone. The complexation with zinc stabilizes the enamine intermediate in aqueous medium. The dissociation of the zinc from the amine group offers a clear route for nucleophilic addition to carbonyl group. The detailed catalytic cycle for Zn-proline catalyzed direct aldol reaction is shown in Scheme 23.
 | ||
| Scheme 23 Catalytic cycle for Zn–proline catalyzed stereoselective aldol reaction in aqueous medium (reprinted with permission from Eur. J. Org. Chem. 2005, 5268 Darbre et al.). | ||
Later Reymond et al. proposed a dual mechanism for the same zinc–proline catalyzed asymmetric aldol reaction in water.76 The zinc–proline complex catalyzes the reaction either by enamine or enolate intermediate (Scheme 24). The Lewis acidic metal centre of the zinc–proline complex is first coordinated to the carbonyl oxygen which is followed by either enamine or enolate mechanism.
 | ||
| Scheme 24 Dual-mechanism for zinc–proline catalyzed enantioselective aldol reaction (reprinted with permission from Chem. Commun. 2006, 1482 Reymond et al.). | ||
Mechanistic studies of aldol reactions of aryl ketones with aryl aldehydes have been carried out using chiral Zn–BINOL complex.44 Here, at first the Et2Zn coordinates with the ligand forming a steady transition state (Scheme 25). The zinc nucleus of this intermediate then gets coordinated to the carbonyl oxygen of both aldehyde and ketone. Here the approach of the zincate enol to benzaldehyde occurs mainly from the Re-face of benzaldehyde and gives β-hydroxy ketones with R-configuration.
 | ||
| Scheme 25 Proposed mechanism for the chiral zinc–BINOL complex catalyzed asymmetric aldol reaction of aryl aldehydes and aryl ketones (reprinted with permission from J. Org. Chem. 2008, 73, 7398 Li et al.). | ||
5. Applications
The aldol reaction is one of the most efficiently used protocols for the stereoselective construction of many naturally occurring polyketides, polyols, sugars and several bioactive molecules. The most effective and atom-economic approach for the synthesis of polyhydroxylated natural products is the aldol reaction. Trost et al. used the dinuclear zinc prolinylphenol catalytic system for the enantioselective total synthesis of (+)-boronolide.32 Boronolide belongs to the class of C-12 lactones having polyhydroxylated side chains. It has been used as a folk medicine in southern Africa. The key step in the synthesis of (+)-boronolide involves the stereoselective Zn-catalyzed aldol reaction between hydroxyacetylfuran and valeraldehyde using the dinuclear Zn–prolinylphenol catalyst 2 (Scheme 26). | ||
| Scheme 26 Dinuclear Zn-catalyzed asymmetric synthesis of (+)-boronolide. | ||
Trost's modified zinc–prolinylphenol catalytic system was also used in the synthesis of β-receptor agonists (−)-arbutamine and (−)-denopamine via stereoselective Henry reaction.55 Asymmetric Henry reaction offers an easy access to arylethanol amines which are used in the treatment of coronary diseases. (−)-Arbutamine was obtained from the nitro-aldol adduct of protected dihydroxylated aromatic aldehyde in two steps (Scheme 27).
 | ||
| Scheme 27 Synthesis of (−)-arbutamine via asymmetric dinuclear Zn-catalyzed Henry reaction. | ||
The (−)-denopamine was prepared from protected monohydroxylated aromatic aldehyde in five steps and the key step involved asymmetric nitro-aldol reaction (Scheme 28).55
 | ||
| Scheme 28 Synthesis of (−)-denopamine via dinuclear Zn-catalyzed enantioselective Henry reaction. | ||
The zinc–proline complex was used for the synthesis of tetrose, pentose and hexoses; but the reaction took longer time for completion.42,43 Depending on the aldehyde substrate, the product could be tetrose, pentose, hexose or smaller sugars (Scheme 29). Being the part of RNA, the pentose sugar synthesis has enormous significance.
 | ||
| Scheme 29 Zn(proline)2-catalyzed synthesis of natural sugars. | ||
Kobayashi's zinc-catalyzed asymmetric Mannich reaction is used for the synthesis of (1R,3R)-N-(3-hydroxy-1-hydroxymethyl-3-phenylpropyl) dodecanamide (HPA-12) (Scheme 30).61 HPA-12 acts as an inhibitor for sphingomyelin synthesis and therefore used as a drug for inhibiting intracellular trafficking of sphingolipids.
 | ||
| Scheme 30 Synthesis of HPA-12 using zinc-catalyzed asymmetric Mannich reaction. | ||
Shibasaki's Zn–Zn-linked BINOL complex 4 offered an efficient practical route for the synthesis of syn-1,2-diols (Scheme 31).38 The diols were further transformed into esters and amides via regioselective rearrangements.
 | ||
| Scheme 31 Synthesis of syn-aldol adducts and transformation to esters and amides by regioselective rearrangements. | ||
6. Scope of enantioselective aldol reactions
The zinc-catalyzed enantioselective aldol reactions in aqueous media help to imitate naturally occurring asymmetric bio-processes, providing a better route for understanding the bio-chemical processes associated with life. It also provides new routes for the development of naturally occurring sugars, polyketides, β-hydroxyamines etc. The reusability of the catalyst and the use of water as the solvent for the reaction make this a green methodology.7. Conclusion
Zinc-catalyzed enantioselective aldol reaction has evolved as a viable route for the construction of C–C bonds. The zinc-complex effectively acts as a Lewis acid and promotes the reaction without the aid of any additional bases or acids. The major advantages of zinc-catalyzed aldol reaction is the very mild reaction condition. The ability of the Zn-catalyst to facilitate the aldol reaction on various substrates such as ynones, methylvinyl ketones etc. is commendable. From the green chemistry point of view, the zinc-catalyzed asymmetric aldol reactions are environmentally friendly as the catalysts can be recycled and the reaction medium is water. However, the reaction has the drawback of long reaction time in many cases. This warrants further research in this area to develop a more efficient zinc-catalyst to perform the reaction in short reaction time.Acknowledgements
The authors are thankful to the Kerala State Council for Science, Technology and Environment (KSCSTE), Trivandrum, India for a research fellowship to APT and research grant (Order No. 341/2013/KSCSTE dated 15.03.2013) to GA. SA and KSS thank the Council of Scientific & Industrial Research (CSIR), India and the University Grants Commission, India for the award of CSIR and UGC fellowships respectively.References
- T. Mukaiyama, Org. React., 1982, 28, 203 CAS
.
- B. Alcaide and P. Almendros, Angew. Chem., Int. Ed., 2003, 42, 858 CrossRef CAS PubMed
- T. Satyanarayana, S. Abraham and H. B. Kagan, Angew. Chem., Int. Ed., 2009, 48, 456 CrossRef CAS PubMed
- X. F. Wu and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141 CrossRef CAS PubMed
- K. Drauz, A. Kleeman and J. Martens, Angew. Chem., Int. Ed., 1982, 21, 584 CrossRef PubMed
- B. M. Trost and C. S. Brindle, Chem. Soc. Rev., 2010, 39, 1600 RSC
- C. Palomo, M. Oiarbide and G. M. Garcia, Chem. Soc. Rev., 2004, 33, 65 RSC
- J. Mlynarski and J. Paradowska, Chem. Soc. Rev., 2008, 37, 1502 RSC
- J. Mlynarski and S. Bas, Chem. Soc. Rev., 2014, 43, 577 RSC
- D. Lowicki, S. Bas and J. Mlynarski, Tetrahedron, 2015, 71, 1339 CrossRef CAS PubMed
- T. J. Brocksom, J. Nakamura, M. L. Ferreira and U. Brocksom, J. Braz. Chem. Soc., 2001, 12, 597 CrossRef CAS
- R. Mahrwald, in Modern Aldol reactions, ed. R. Mahrwald, Wiley, 2004, vol. 2, p. 1 Search PubMed
- Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893 RSC
- X. F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084 RSC
- Z. Xie, Y. Cai, H. Hu, C. Lin, J. Jiang, Z. Chen, L. Wang and Y. Pan, Org. Lett., 2013, 15, 4600 CrossRef CAS PubMed
- H. J. Cristau, B. Chabaud, R. Labaudiniere and H. Christol, J. Org. Chem., 1986, 51, 875 CrossRef CAS
- M. T. Lan, W. Y. Wu, S. H. Huang and K. L. Luo, RSC Adv., 2011, 1, 1751 RSC
- A. Correa, M. Carril and C. Bolms, Chem.–Eur. J., 2008, 14, 10919 CrossRef CAS PubMed
- M. A. Fernández-Ibánez, B. Maciá, A. J. Minnaard and B. L. Feringa, Angew. Chem., Int. Ed., 2008, 47, 1317 CrossRef PubMed
- P. G. Cozzi, A. Mignogna and P. Vicennati, Adv. Synth. Catal., 2008, 350, 975 CrossRef CAS PubMed
- T. Hama, S. Ge and J. F. Hartwig, J. Org. Chem., 2013, 78, 8250 CrossRef CAS PubMed
- A. Krasovskiy, C. Duplaces and B. H. Lipshutz, J. Am. Chem. Soc., 2009, 131, 15592 CrossRef CAS PubMed
- Y. Mori and M. Seki, Synlett, 2005, 2233 CAS
- H. Dong, M. Shen, J. E. Redford, B. J. Stokes, A. L. Pumphrey and T. G. Driver, Org. Lett., 2007, 9, 5191 CrossRef CAS PubMed
- P. G. Cozzi and E. Rivalta, Angew. Chem., Int. Ed., 2005, 44, 3600 CrossRef CAS PubMed
- B. Alcaide and P. Almendros, Eur. J. Org. Chem., 2002, 1595 CrossRef CAS
- K. Irie and K. Watanabe, Bull. Chem. Soc. Jpn., 1980, 53, 1366 CrossRef CAS
- K. Watanabe, Y. Yamada and K. Goto, Bull. Chem. Soc. Jpn., 1985, 58, 1401 CrossRef CAS
- B. M. Trost and H. Ito, J. Am. Chem. Soc., 2000, 122, 12003 CrossRef CAS
- B. M. Trost, H. Ito and E. R. Silcoff, J. Am. Chem. Soc., 2001, 123, 3367 CrossRef CAS
- B. M. Trost and V. S. C. Yeh, Org. Lett., 2002, 4, 3513 CrossRef CAS PubMed
- B. M. Trost, E. R. Silcoff and H. Ito, Org. Lett., 2001, 3, 2497 CrossRef CAS PubMed
- B. M. Trost, A. Fettes and B. T. Shiremen, J. Am. Chem. Soc., 2004, 126, 2660 CrossRef CAS PubMed
- B. M. Trost, S. Shin and J. A. Sclafani, J. Am. Chem. Soc., 2005, 127, 8602 CrossRef CAS PubMed
- N. Yoshikawa, N. Kumagai, S. Matsunaga, G. Moll, T. Ohshima, T. Suzuki and M. Shibasaki, J. Am. Chem. Soc., 2001, 123, 2466 CrossRef CAS
- N. Kumagai, S. Matsunaga, T. Kinoshita, S. Harada, S. Okada, S. Sakamoto, K. Yamaguchi and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 2169 CrossRef CAS PubMed
- N. Kumagai, S. Matsunaga, N. Yoshikawa, T. Ohshima and M. Shibasaki, Org. Lett., 2001, 3, 1539 CrossRef CAS PubMed
- T. Darbre, C. Dubs, E. Rusanov and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2002, 3284 CrossRef CAS
- T. Darbre and M. Machuquiero, Chem. Commun., 2003, 1090 RSC
- R. Fernandez-Lopez, J. Kofoed, M. Machuqueiro and T. Darbre, Eur. J. Org. Chem., 2005, 5268 CrossRef CAS PubMed
- J. Kofoed, M. Machuqueiro, J.-L. Reymond and T. Darbre, Chem. Commun., 2004, 1540 RSC
- J. Kofoed, J. L. Reymond and T. Darbre, Org. Biomol. Chem., 2005, 3, 1850 CAS
- H. Li, C. S. Da, Y. H. Xiao, X. Li and Y. N. Su, J. Org. Chem., 2008, 73, 7398 CrossRef CAS PubMed
-
(a) D. Voet, in Biochemistry, eds. D. Voet and J. G. Voet, John Wiley and Sons, New York, 1990 Search PubMed
- S. Itoh, M. Kitamura, Y. Yamada and S. Aoki, Chem.–Eur. J., 2009, 15, 10570 CrossRef CAS PubMed
-
(a) J. Jankowska and J. Mlynarski, J. Org. Chem., 2006, 71, 1317 CrossRef CAS PubMed
- G. Chen, X. Fu, C. Li, C. Wu and Q. Miao, J. Organomet. Chem., 2012, 702, 19 CrossRef CAS PubMed
- J. Paradowska, M. Stodulski and J. Mlynarski, Adv. Synth. Catal., 2007, 349, 1041 CrossRef CAS PubMed
- L. ZhiJin, M. HaiBo, Z. GuangQian, H. JianLin and P. Yi, Sci. China: Chem., 2010, 53, 2291 CrossRef
- L. ZhiJin, M. HaiBo, J. Han and Y. Pan, Chem. Biol. Drug Des., 2010, 76, 181 Search PubMed
-
(a) L. Henry, C. R. Hebd. Seances Acad. Sci., 1895, 120, 1265 CAS
- B. M. Trost and V. S. C. Yeh, Angew. Chem., Int. Ed., 2002, 41, 861 CrossRef CAS
- C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS PubMed
- B. M. Trost, V. S. C. Yeh, H. Ito and N. Bremeyer, Org. Lett., 2002, 4, 2621 CrossRef CAS PubMed
- B. M. Trost and D. W. Lupton, Org. Lett., 2007, 9, 2023 CrossRef CAS PubMed
- J. Gao and A. E. Martell, Org. Biomol. Chem., 2003, 1, 2801 CAS
- C. Palomo, M. Ocarbide and A. Laso, Angew. Chem., Int. Ed., 2005, 44, 3881 CrossRef CAS PubMed
- C. Palomo, M. Ocarbide, R. Halder, A. Laso and R. Lopez, Angew. Chem., Int. Ed., 2006, 45, 117 CrossRef CAS PubMed
- A. Cὀrdova, Acc. Chem. Res., 2004, 37, 102 CrossRef PubMed
- S. Kobayashi, T. Hamada and K. Manabe, J. Am. Chem. Soc., 2002, 124, 5640 CrossRef CAS PubMed
- B. M. Trost and L. R. Terell, J. Am. Chem. Soc., 2003, 125, 338 CrossRef CAS PubMed
- S. Matsunaga, N. Kumagai, S. Harada and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 4712 CrossRef CAS PubMed
- S. Matsunaga, T. Yoshida, H. Morimoto, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 8777 CrossRef CAS PubMed
- B. M. Trost, J. Jaratjaroonphong and V. Reutrakul, J. Am. Chem. Soc., 2006, 128, 2778 CrossRef CAS PubMed
- Z. Yuan, L. Mei, Y. Wei, M. Shi, P. V. Kattumuri, P. McDowell and G. Li, Org. Biomol. Chem., 2012, 10, 2509 CAS
- Z. L. Yuan, Y. Wei and M. Shi, Chin. J. Chem., 2010, 28, 1709 CrossRef CAS PubMed
- H. Schofield, J. Fluorine Chem., 1999, 100, 7 CrossRef CAS
- N. Krause and A. Hoffmann-Röder, Synthesis, 2001, 171 CrossRef CAS
- S. Harada, N. Kumagai, T. Kinoshita, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2003, 125, 2582 CrossRef CAS PubMed
- B. M. Trost and K. Hirano, Angew. Chem., Int. Ed., 2012, 51, 6480 CrossRef CAS PubMed
-
(a) R. A. Sheldon, in Green Chemistry and Catalysis, ed. R. A. Sheldon, I. Arends and U. Hanefeld, Wiley-VCH, Weinheim, 1st edn, 2007 Search PubMed
- R. B. N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559 RSC
- J. Paradowska, M. Stodulski and J. Mlynarski, Angew. Chem., Int. Ed., 2009, 48, 4288 CrossRef CAS PubMed
- T. D. Machajewski and C. H. Wong, Angew. Chem., Int. Ed., 2000, 39, 1352 CrossRef CAS
- J. Kofoed, T. Darbre and J. L. Reymond, Chem. Commun., 2006, 1482 RSC
| This journal is © The Royal Society of Chemistry 2015 |