
A stereocontrolled synthesis of Hagen's gland lactones via iterative proline catalyzed α-aminoxylation and oxa-Michael addition reactions†
Shruti Vandana Kauloorkara,
Vishwajeet Jhaa,
Ganesh Jogdandb and
Pradeep Kumar*a
aDivision of Organic Chemistry, CSIR-NCL (National Chemical Laboratory), Pune 411008, India. E-mail: pk.tripathi@ncl.res.in; Tel: +9102025902627
bCentral NMR Facility, CSIR-NCL (National Chemical Laboratory), Pune 411008, India
First published on 7th July 2015
Abstract
A simple and efficient synthesis of Hagen's gland lactones was achieved using a sequential α-aminoxylation/oxa-Michael approach in a highly diastereoselective manner with assignment of relative configurations. This method was found to be applicable to the synthesis of various other isomers of Hagen's gland lactones.
Introduction
Hagen's glands (Fig. 1) also known as pygidial glands (located near the abdominal tips) of braconid wasps, D. longicaudata (Ashmead), D. tryoni (Cameron) and Fopius arisanus (Biosteres), are found to contain fragrance rich lactones. This was first observed by Hagen (1953) and Buckingham (1975) who also made an effort to study the significance of these secretions in pest management to control the fruitfly population in Hawaii and Eastern Queensland, especially for the Queensland fruitfly. Bactrocera tryoni is known to be an aggressive pest with a wide host range.1 Williams et al. suggested the presence of two bicyclic lactones and experimentally characterized these bicyclic lactones by NMR studies using Karplus based calculations.1c Kitching et al. have determined the absolute stereochemistry of these lactones through synthesis which employs an interesting route that uses 1,3-diol approach followed by PdCl2-catalyzed oxy carbonylation–lactonization reaction.2a | ||
| Fig. 1 Hagen's gland lactones (2 & 4) and their epimers (1 & 3). | ||
Considering their possible role in pest management strategies, several authors have reported the synthesis of these lactones either targeting the natural isomer or its epimer. Chiral pool approaches have been employed for the synthesis of Hagen's gland lactones from carbohydrates,3,4 chiral glycidols,5 and lactones derived from carbohydrates such as mannofuranolactone6 and D-glucono-δ-lactone.7a,b Very recently, Lepore et al. described an enantioselective synthesis of Hagen's gland lactones from 2,3-allenols.7c In yet another report Gharpure et al. made use of synthetic intermediates like cyclopropanes (DAC) for the synthesis of target lactones.8
During last decade, there has been growing interest in the use of small organic molecules to catalyze reactions in a stereoselective manner in organic synthesis. Proline is among the most successful secondary amine based organocatalysts which have been widely employed in several organic transformations.9
As a part of our research interest in developing new methodologies and their subsequent application to bioactive compounds,10 we have recently developed an iterative approach to enantiopure synthesis of syn and anti-1,3-polyols based on proline catalyzed sequential α-aminoxylation, followed by Horner–Wadsworth–Emmons olefination of aldehydes at ambient temperature.11a This method has several advantages over the most widely used method to prepare 1,3-polyols in an iterative fashion. We have earlier reported the synthesis of various lactones using 1,3-polyol approach.11b However the construction of bridged framework containing THF ring systems using the same remains unexplored. We now report the application of this methodology along with a highly diastereoselective oxa-Michael addition reaction in the efficient synthesis of substituted tetrahydrofuro [3,2-b]furan-2(3H)-one derivatives (Hagen's gland lactones).
Results and discussion
As per the retrosynthetic scheme as delineated in Scheme 1, the Hagen's gland lactones could be synthesized from the skipped 1,3-diol fragment A. We envisioned that A could be derived from γ-hydroxy ester moiety B, a common intermediate which in turn could be obtained via iterative sequential α-aminoxylation and Horner–Wadsworth–Emmons olefination of aldehyde C.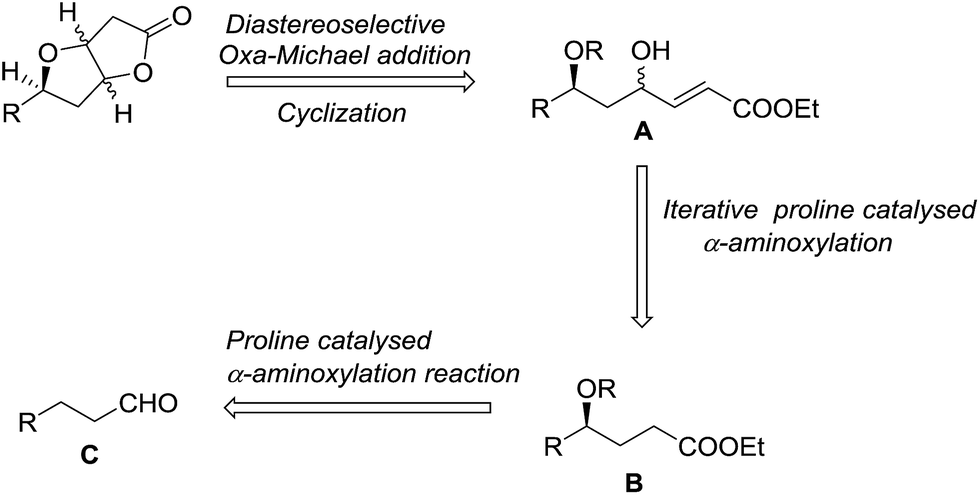 | ||
| Scheme 1 Retrosynthetic route to the synthesis of Hagen's gland lactones. | ||
As shown in Scheme 2, the synthesis of the target lactones commenced with the commercially available hexanal 5a which on sequential α-aminoxylation using nitroso benzene as the oxygen source and L-proline as catalyst and subsequent HWE olefination using triethylphosphonoacetate, followed by hydrogenation using a catalytic amount of Pd/C, furnished the γ-hydroxy ester 6a. Thus, in two steps and one column purification 6a was obtained in 65% yield and 94% ee.11c Similarly compound 6b was obtained from 5b in 65% yield and 98% ee. Protection of the free hydroxyl group of 6a & 6b as its TBS ether gave 7a & 7b in 92% yield respectively. The TBS protected hydroxyester 7a was then reduced using DIBAL-H in toluene at −78 °C to furnish an aldehyde.
 | ||
| Scheme 2 Synthesis of Hagen's gland lactones. | ||
Crude aldehyde was further subjected to α-aminoxylation reaction using L-proline as a catalyst followed by HWE-olefination to yield syn TBS protected γ-hydroxy ester 8a in good diastereomeric excess (dr ratio 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5).12 Using the same procedure 8b was obtained from 7b in 71% yield (dr ratio 96:4). With syn-1,3-diol 8a in hand we proceeded to the synthesis of Hagen's gland lactones using oxa-Michael addition. The key steps involved the fluoride-mediated cleavage of a silyl protecting group using TBAF in THF followed by lactonization with catalytic amount of HCl (pH ∼ 3 in toluene). At this stage we could observe the formation of two products 1 & 2 (ratio 5:1).13 In a similar way compounds 3 and 4 were obtained from 8b using oxa-Michael addition followed by lactonization.
:5).12 Using the same procedure 8b was obtained from 7b in 71% yield (dr ratio 96:4). With syn-1,3-diol 8a in hand we proceeded to the synthesis of Hagen's gland lactones using oxa-Michael addition. The key steps involved the fluoride-mediated cleavage of a silyl protecting group using TBAF in THF followed by lactonization with catalytic amount of HCl (pH ∼ 3 in toluene). At this stage we could observe the formation of two products 1 & 2 (ratio 5:1).13 In a similar way compounds 3 and 4 were obtained from 8b using oxa-Michael addition followed by lactonization.
Taking into consideration this observation we considered it worthwhile to study the stereochemistry of both the products which was confirmed using detailed 1D and 2D-NMR techniques.
For compound 1, proton H3a shows nOe correlation with proton H6a indicating syn stereochemistry at the bridgehead of the substituted tetrahydrofuro[3,2-b]furan-2(3H)-one. H3a also shows nOe correlation with proton H5, which confirms the syn relative stereochemistry between these three protons as shown in the pictorial representation of the compound in the Fig. 2 (see ESI† for spectra).
 | ||
| Fig. 2 Pictorial representation of both cis and trans nOe correlations. | ||
In case of compound 2, the H5 proton shows nOe correlation with H6α′ while H6a shows nOe correlation with H6β′. These results show that the H6a and H5 methine protons show nOe correlation with different protons of the furyl methylene indicating anti relative stereochemistry between H5 and H6a as shown in the pictorial representation in Fig. 2 (see ESI† for spectra).
This result motivated us to study the stereoselection of both oxa-Michael and cyclization reactions very closely. The reproducibility of the strategy and high yielding steps efficiently allowed us to quickly synthesize 1,3-syn-diol 8a which was subjected to simultaneous desilylation/oxa-Michael reaction. Instead of going further for cyclization at this stage, we quenched the reaction mixture using saturated ammonium chloride solution to get the oxa-Michael product 9 (Scheme 3). Preliminary examination using thin layer chromatography showed the presence of only one product. 1H and 13C NMR (see ESI†) did not show the formation of other diastereomer and revealed that the oxa-Michael addition reaction proceeded in a highly diastereoselective manner. The stereochemistry of compound 9 was confirmed using detailed 1D and 2D NMR techniques (see ESI† for spectra).
 | ||
| Scheme 3 Diastereoselective oxa-Michael addition reaction of 8a. | ||
It was observed that in compound 9, H4 and H1 shows nOe correlations indicating syn relative stereochemistry14 while none of them shows nOe correlations with H3 indicating anti relative stereochemistry with H3 as shown in the pictorial representation in Fig. 3. The possible reason for the formation of mixture of diastereomers in the cyclization step could be attributed to the epimerisation of either of the two protons (H3 or H4) in the presence of HCl (pH ∼ 3) under reflux conditions, leading to cyclization with both the ring junction protons syn to each other. To prevent the racemization and to check the feasibility of cyclization of 9 without epimerisation, we further carried out reaction using p-TSA in toluene both at room temperature and under reflux conditions. As anticipated, the cyclization reaction proved to be a total failure as it gave only the starting material back (Scheme 4).
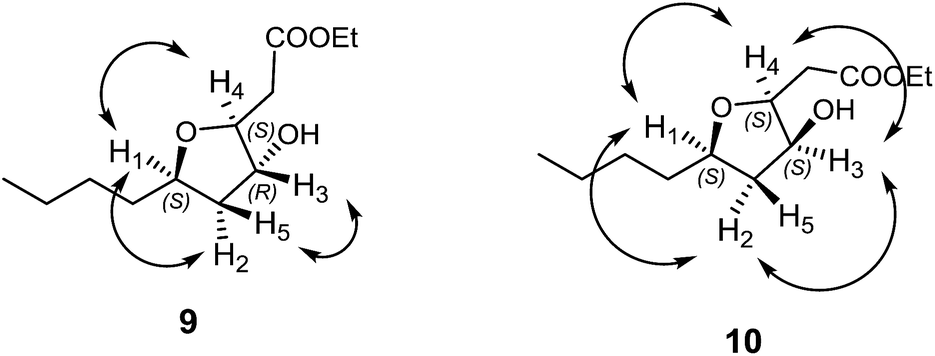 | ||
| Fig. 3 nOe correlations for compounds 9 and 10. | ||
 | ||
| Scheme 4 Lactonization reaction. | ||
In order to rationalise our findings, we planned to test the devised strategy by synthesizing 1,3-anti-diol as an intermediate. For this purpose, we started with previously synthesized protected γ-hydroxy ester 7a which was reduced using DIBAL-H in toluene at −78 °C to furnish corresponding aldehyde. Crude aldehyde was further subjected to α-aminoxylation/HWE olefination reaction using D-proline as a catalyst to obtain 1,3-anti-diol 8c with an excellent dr ratio (97:3).13 To test the formation of diastereomeric mixture one-pot oxa-Michael/lactonization was performed on the diol. In this case we observed the formation of only one product as characterized by 13C-NMR which was an entirely different result when compared to the syn-diol product (Scheme 5).
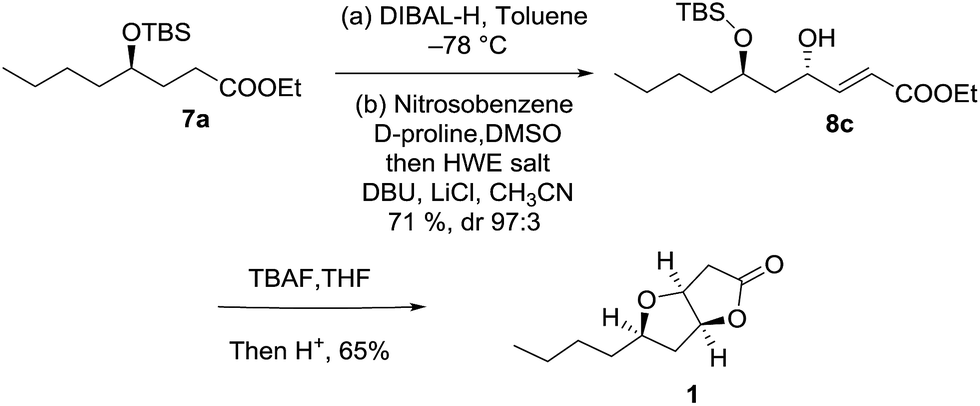 | ||
| Scheme 5 Synthesis of epi-Hagen's gland lactone. | ||
This observation was further substantiated and proved by isolating compound 10 and examining the course of reaction by its treatment under various acidic conditions. Towards this end, compound 8c was subjected to a concomitant desilylation and oxa-Michael with TBAF in THF for 3 h to obtain 10 in 85% yield as shown in Scheme 6. Characterization of the oxa-Michael adduct 10 was carried out by 1D and 2D NMR techniques.
 | ||
| Scheme 6 Synthesis of compound 10. | ||
For compound 10, H3 proton shows nOe correlation with both methine protons H1 and H4 indicating syn stereochemistry among them. The relative stereochemistry was also confirmed with the help of methylene group which shows two different signals for two protons (H2 and H5). The H1 and H3 methine protons show nOe correlations only with H2 proton, but it does not show any correlation with H5 proton indicating all the three methine protons (H1, H3 and H4) being syn to each other as shown in the pictorial representation in Fig. 3.
After confirming the stereochemistry of compound 10, it was initially subjected to cyclization using p-TSA at rt to give compound 1 as sole product (Scheme 7). We then examined the epimerisation using reflux conditions in the presence of p-TSA, when no epimerisation occurred we then tried using conc. HCl at both rt and under reflux conditions. Interestingly, no epimerisation was observed and cyclization was smooth leading to the desired product 1 in excellent yield.
 | ||
| Scheme 7 Lactonization reaction. | ||
This could be due to the syn stereochemistry of the intermediate making cyclization more facile than the epimerisation at C-3 centre. To check the reproducibility and improve the confidence in the stereochemical outcome by the above methods, we thought of extrapolating the strategy to the synthesis of 3 and 4 isolated from D. krausii. Both the compounds could easily be synthesized to obtain a separable mixture of cis and trans isomers from the corresponding aldehyde octanal 5b by subjecting it to similar set of reaction conditions as described in Scheme 2. Thus, epi-Hagen's gland lactone 1 was obtained in overall ∼28% yield and Hagen's gland lactone 2 in ∼4.5% yield starting from cheap and easily available aldehydes in 4 steps only.
Conclusions
In conclusion, we have developed a new, efficient and organocatalytic protocol to Hagen's gland lactones using a proline catalyzed α-aminoxylation and consequent oxa-Michael reactions. We believe that this approach would permit maximum variability in the product structure and can be extended to the synthesis of other stereoisomers and synthetic analogues. The synthesis reported uses mild reaction conditions (at room temperature, air and moisture are tolerated), both enantiopure forms of proline are commercially available, Thus by using the suitable catalyst, desirable stereocenters can be obtained (highly stereo divergent). A short reaction sequence with a stereochemical assignment has made this approach amenable to similar natural products. Currently, studies are in progress toward this goal.Experimental section
Ethyl (R)-4-hydroxydecanoate (6b)
:EtOAc (85:15) as eluent to give ethyl (R)-4-hydroxydecanoate 6b as a colourless liquid (2.19 g, yield 65%). [α]25D: +1.17 (c 1.5, CHCl3), IR (CHCl3, cm−1): νmax 3432, 2934, 1718. 1H NMR (200 MHz, CDCl3) δ 4.13 (q, J = 7.2 Hz, 2H), 3.67–3.52 (m, 1H), 2.50–2.39 (m, 2H), 1.99–1.61 (m, 4H), 1.56–1.29 (m, 8H), 1.29–1.22 (m, 3H), 0.92 (d, J = 4.8 Hz, 3H) ppm. 13C NMR (126 MHz, CDCl3): δ 174.1, 80.9, 71.0, 60.2, 37.4, 32.1, 30.7, 29.5, 25.5, 22.5, 14.0, 13.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C12H24O3Na 239.1618; found 239.1614.HPLC: Kromasil 5-Amycoat (250 × 4.6 mm) (2-propanol:petroleum ether = 10:90), flow rate 0.5 mL min−1. Retention time (min): 78.483 (minor) and 80.708 (major). The racemic standard was prepared in the same way using DL-proline as a catalyst. ee > 98%.
Ethyl (R)-4-((tert-butyldimethylsilyl)oxy)decanoate (7b)
To an ice-cold stirred solution of 6b (1.70 g, 7.87 mmol) in DMF (10 mL) were added imidazole (1.00 g, 15.74 mmol) and TBSCl (1.77 g, 11.80 mmol) at 0 °C. The resulting mixture was stirred for 1 h at rt before H2O (20 mL) was added. The aqueous layer was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. Silica gel column chromatography using petroleum ether:ethyl acetate: (95:05) of the crude product gave TBS ether 7b as a colorless liquid (2.39 g, yield 92%). [α]25D: −7.08 (c 1.6, CHCl3), IR (CHCl3, cm−1): νmax 2856, 1726. 1H NMR (200 MHz, CDCl3) δ 4.12 (q, J = 7.1 Hz, 2H), 3.75–3.59 (m, 1H), 2.40–2.29 (m, 2H), 1.82–1.63 (m, 2H), 1.47–1.34 (m, 2H), 1.30–1.22 (m, 11H), 0.88 (m, 12H), 0.04 (s, 6H) ppm. 13C NMR (101 MHz, CDCl3) δ 174.0, 71.2, 60.2, 37.0, 31.8, 31.7, 30.1, 29.5, 25.9, 25.1, 22.6, 18.1, 14.2, 14.1, −4.4, −4.6 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C18H38O3NaSi 353.2482; found 353.2472.
Ethyl (4R,6R,E)-6-((tert-butyldimethylsilyl)oxy)-4-hydroxydec-2-enoate (8a)
To a solution of ethyl ester 7a (1.0 g, 4.63 mmol) in CH2Cl2 (6 mL), was added DIBAL-H (2.5 mL 2.3 M solution in toluene, 5.09 mmol) at −78 °C under argon atmosphere. The reaction was stirred at this temperature for 40 min. Then a solution of tartaric acid (2.5 mL) was added. The resulting mixture was stirred for 15 min and the organic layer was separated. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL), the combined organic layers were dried (Na2SO4), filtered and evaporated under reduced pressure to give aldehyde as a colourless liquid, which was directly used in the next step without further purification. Following the general procedure for α-aminoxylation (L-proline as a catalyst) 8a was obtained as a crude product (∼95% diastereomeric excess) and was purified by flash column chromatography using petroleum ether:ethyl acetate (9:1) to furnish pure diol 8a as a colorless liquid (0.80 g, yield 71%). [α]25D: −15.76 (c 0.6, CHCl3), IR (CHCl3, cm−1): νmax 3436, 2967, 1218. 1H NMR (200 MHz, CDCl3) δ 6.92 (dd, J = 4.4, 15.6 Hz, 1H), 6.10 (dd, J = 1.8, 15.6 Hz, 1H), 4.46 (m, 1H), 4.19 (q, J = 7.1 Hz, 2H), 3.96 (m, 1H), 1.76–1.50 (m, 4H), 1.27–1.23 (m, 7H), 0.94–0.90 (m, 12H), 0.16–0.08 (m, 6H) ppm. 13C NMR (126 MHz, CDCl3) δ 166.7, 149.7, 119.8, 73.3, 70.4, 60.3, 41.9, 37.7, 26.8, 25.8, 22.8, 17.9, 14.2, 14.0, −4.0, −4.8 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C18H36O4NaSi 367.2275; found 367.2275.
HPLC: Kromasil RP-18 (150 × 4.6 mm) (acetonitrile:H2O = 90:10), flow rate 1.0 mL min−1, (λ = 210 nm). Retention time (min): 7.31 (major) and 7.89 (minor), (dr 95:5).
Ethyl (4S,6R,E)-6-((tert-butyldimethylsilyl)oxy)-4-hydroxydec-2-enoate (8c)
The above procedure was followed using D-Proline as a catalyst (0.79 g, yield 70%) [α]25D: −7.5 (c 0.4, CHCl3), IR (CHCl3, cm−1): νmax 3430, 2934, 1718. 1H NMR (200 MHz, CDCl3) δ 6.92 (dd, J = 4.2, 15.6 Hz, 1H), 6.11 (dd, J = 1.9, 15.5 Hz, 1H), 4.64 (dtd, J = 1.8, 4.2, 8.2 Hz, 1H), 4.20 (q, J = 7.1 Hz, 2H), 4.05–3.94 (m, 1H), 1.74–1.65 (m, 2H), 1.29 (t, J = 7.1 Hz, 9H), 0.90 (m, 12H), 0.09 (d, J = 1.4 Hz, 6H) ppm. 13C NMR (101 MHz, CDCl3): δ 166.7, 150.4, 119.8, 71.5, 68.0, 60.3, 40.5, 35.7, 27.8, 25.8, 22.7, 17.9, 14.2, 14.0, −4.5, −4.8 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C18H36O4NaSi 367.2273; found 367.2275.HPLC: Kromasil RP-18 (150 × 4.6 mm) (acetonitrile:H2O = 90:10), flow rate 1.0 mL min−1, (λ = 210 nm). Retention time (min): 6.91 (major) and 7.43 (minor), dr 3:97.
Ethyl (4R,6R,E)-6-((tert-butyldimethylsilyl)oxy)-4-hydroxydodec-2-enoate (8b)
(0.80 g, yield 70%) [α]25D: −6.49 (c 1.8, CHCl3), IR (CHCl3, cm−1): νmax 3450, 2920. 1H NMR (200 MHz, CDCl3) δ 6.91 (dd, J = 4.4, 15.6 Hz, 1H), 6.10 (dd, J = 1.9, 15.5 Hz, 1H), 4.24–4.09 (m, 2H), 4.07–3.82 (m, 1H), 3.82–3.51 (m, 1H), 1.63–1.52 (m, 4H), 1.27 (d, J = 4.5 Hz, 11H), 0.92–0.88 (m, 12H), 0.12 (d, J = 2.5 Hz, 6H) ppm. 13C NMR (126 MHz, CDCl3) δ 166.7, 149.7, 119.8, 73.3, 70.4, 60.3, 42.0, 38.0, 31.8, 29.4, 25.8, 24.6, 22.5, 17.9, 14.2, 14.0, −4.0, −4.7 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C20H40O4NaSi 395.2588; found 395.2578.HPLC: Kromasil RP-18 (150 × 4.6 mm) (acetonitrile:H2O = 90:10), flow rate 1.0 mL min−1, (λ = 210 nm). Retention time (min): 10.61 (major) and 11.61 (minor), dr 96:4.
Ethyl 2-((2S,3R,5R)-5-butyl-3-hydroxytetrahydrofuran-2-yl)acetate (9)
The solution of 8a (0.25 g, 0.92 mmol) was treated with TBAF (0.5 mL, 1.8 mmol) in THF (3 mL) at 0 °C. The reaction mixture was stirred for 3 h and quenched with saturated ammonium chloride solution (1 mL) and extracted with ethyl acetate (3 × 3 mL). The combined organic layers were washed with brine, dried over anhyd. Na2SO4 and concentrated under reduced pressure to give a crude product. Silica gel column chromatography using petroleum ether:ethyl acetate: (8:2) of the crude product gave oxa-Michael product 9 as a colorless liquid (0.14 g, yield 85%).
[α]25D: +1.3 (c 0.3, CHCl3). IR (CHCl3, cm−1): νmax 3463, 2931, 1764. 1H NMR (400 MHz, CDCl3) δ 4.18 (d, J = 7.0 Hz, 2H), 4.14–4.10 (m, 1H), 4.06 (dd, J = 6.1, 9.2 Hz, 1H), 3.95 (td, J = 4.6, 9.4 Hz, 1H), 2.80 (dd, J = 5.0, 16.3 Hz, 1H), 2.51 (dd, J = 9.2, 16.5 Hz, 1H), 2.35 (t, J = 7.6 Hz, 1H), 2.01–1.96 (m, 1H), 1.82–1.75 (m, 1H), 1.49–1.42 (m, 2H), 1.33–1.26 (m, 6H), 0.91–0.87 (m, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 172.2, 82.3, 78.4, 77.2, 60.9, 40.3, 38.8, 35.2, 28.1, 22.7, 14.1, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C12H22O4Na 253.1410; found 253.1411.
Ethyl 2-((2S,3S,5R)-5-hexyl-3-hydroxytetrahydrofuran-2-yl)acetate (10)
10 was prepared using same procedure as described for 9. (0.14 g, yield 85%). [α]25D: −8.4 (c 1.0, CHCl3), IR (CHCl3, cm−1): νmax 3463, 2931, 1764. 1H NMR (700 MHz, CDCl3) δ 4.13–4.05 (m, 3H), 3.98–3.93 (m, 1H), 3.93–3.88 (m, 1H), 2.66 (dd, J = 5.3, 16.3 Hz, 1H), 2.45–2.41 (m, 1H), 2.30 (td, J = 6.7, 13.0 Hz, 1H), 1.58 (dd, J = 4.9, 7.9 Hz, 1H), 1.44–1.41 (m, 2H), 1.30–1.18 (m, 7H), 0.82–0.78 (m, 3H) ppm. 13C NMR (176 MHz, CDCl3) δ 172.4, 79.8, 77.7, 76.8, 60.9, 40.2, 38.6, 35.8, 28.0, 25.7, 22.6, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C12H22O4Na 253.1410; found 253.1411.(3aS,5R,6aS)-5-Butyltetrahydrofuro[3,2-b]furan-2(3H)-one (1)
:ethyl acetate: (85:15) afforded 1 as a syrupy liquid (0.047 g, 61%). Further chromatography with Pet ether:ethylacetate: (85:15) gave the other isomer 2 as a syrupy liquid (0.009 g, 12.2%). [α]25D: −53.39 (c 0.8, CHCl3) [lit.2b [α]D = −53.9], 1H NMR (400 MHz, CDCl3) δ 5.03–5.00 (m, 1H), 4.53–4.50 (m, 1H), 3.97–3.91 (m, 1H), 2.73 (d, J = 3.3 Hz, 2H), 2.46–2.39 (m, 1H), 1.91–1.86 (m, 1H), 1.70–1.65 (m, 1H), 1.60–1.54 (m, 1H), 1.37–1.30 (m, 4H), 0.92–0.89 (m, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 175.5, 84.7, 80.3, 78.2, 38.3, 36.6, 35.2, 28.2, 22.6, 13.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C10H16O3Na 207.0991; found 207.0992.(3aR,5R,6aR)-5-Butyltetrahydrofuro[3,2-b]furan-2(3H)-one (2)
[α]25D: +47.22 (c 0.3, CHCl3) [lit.4 [α]D = +50.9 (c = 1.0, CHCl3], 1H NMR (700 MHz, CDCl3) δ 5.12 (t, J = 4.7 Hz, 1H), 4.83–4.79 (m, 1H), 4.07 (dt, J = 6.0, 10.7 Hz, 1H), 2.76 (dd, J = 6.7, 18.7 Hz, 1H), 2.65 (d, J = 18.9 Hz, 1H), 2.38 (dd, J = 4.5, 13.9 Hz, 1H), 1.69–1.65 (m, 1H), 1.56–1.43 (m, 2H), 1.39–1.28 (m, 4H), 0.91 (t, J = 7.1 Hz, 3H) ppm. 13C NMR (176 MHz, CDCl3) δ 176.0, 84.9, 78.3, 77.3, 38.8, 36.6, 34.4, 28.2, 22.6, 13.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C10H16O3Na 207.0991; found 207.0991.(3aS,5R,6aS)-5-Hexyltetrahydrofuro[3,2-b]furan-2(3H)-one (3)
(0.046 g, 60%). [α]25D: −39.62 (c 0.8, CHCl3) [lit.2b [α]D = −41.0], 1H NMR (500 MHz, CDCl3) δ 5.01 (ddd, J = 2.3, 4.6, 6.9 Hz, 1H), 4.53–4.49 (m, 1H), 3.94 (td, J = 7.0, 13.8 Hz, 1H), 2.42 (td, J = 7.2, 14.2 Hz, 2H), 1.89 (ddd, J = 2.3, 7.9, 14.2 Hz, 2H), 1.60–1.50 (m, 4H), 1.37–1.30 (m, 6H), 0.90 (s, 3H) ppm. 13C NMR (126 MHz, CDCl3) δ 175.5, 84.7, 80.4, 78.2, 38.3, 36.4, 35.2, 29.7, 29.6, 28.2, 22.6, 14.0 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C12H20O3Na 235.1302; found 235.1302.(3aR,5R,6aR)-5-Hexyltetrahydrofuro[3,2-b]furan-2(3H)-one (4)
(0.009 g, 12%). [α]25D: +42.78 (c 0.5, CHCl3) [lit.6 [α]D = +50.6 (c 1.0, CHCl3)], 1H NMR (500 MHz, CDCl3) δ 5.13 (t, J = 4.7 Hz, 1H), 4.83 (t, J = 5.5 Hz, 1H), 4.08 (dt, J = 6.1, 10.8 Hz, 1H), 2.77 (dd, J = 6.6, 18.8 Hz, 1H), 2.66 (d, J = 18.9 Hz, 1H), 2.39 (dd, J = 4.6, 13.7 Hz, 2H), 1.73–1.58 (m, 4H), 1.54–1.43 (m, 2H), 1.38–1.32 (m, 4H), 0.93–0.91 (m, 3H) ppm. 13C NMR (126 MHz, CDCl3) δ 175.5, 84.7, 78.2, 77.3, 38.2, 36.3, 35.2, 31.9, 29.6, 29.3, 22.5, 13.9 ppm. HRMS (ESI) m/z: [M + Na]+ calcd for C12H20O3Na 235.1304; found 235.1305.Acknowledgements
S. V. K. thanks UGC New Delhi for fellowship. We thank Ms S. Kunte for HPLC analysis. Financial support from CSIR, New Delhi 12th Five year Plan Project (Grant No. CSC-0108) is gratefully acknowledged.Notes and references
- (a) K. L. Hagen, Proc. Hawaii. Entomol. Soc., 1953, 15, 115 Search PubMed; (b) G. R. Buckingham, Ann. Entomol. Soc. Am., 1968, 61, 233 CrossRef PubMed; (c) H. J. Williams, M. Wong, R. A. Wharton and S. B. Vinson, J. Chem. Ecol., 1988, 14, 1727 CrossRef CAS PubMed.
- (a) G. C. Paddon-Jones, C. J. Moore, D. J. Brecknell, W. A. Konig and W. Kitching, Tetrahedron Lett., 1997, 38, 3479 CrossRef CAS; (b) G. C. Paddon-Jones, C. S. P. McErlean, P. P. Hayes, C. J. Moore, W. A. Konig and W. Kitching, J. Org. Chem., 2001, 66, 7487 CrossRef CAS PubMed.
- (a) H. B. Mereyala and R. R. Gadikota, Chem. Lett., 1999, 273 CrossRef CAS; (b) H. B. Mereyala and R. R. Gadikota, Tetrahedron: Asymmetry, 2000, 11, 743 CrossRef CAS; (c) H. B. Mereyala, R. R. Gadikota, K. S. Sunder and S. Shailaja, Tetrahedron, 2000, 56, 3021 CrossRef CAS.
- E. Paz-Morales, M. Ruth and F. Sartillo-Piscil, Carbohydr. Res., 2009, 344, 1123 CrossRef CAS PubMed.
- D. Agrawal, V. Sriramurthy and V. K. Yadav, Tetrahedron Lett., 2006, 47, 7615 CrossRef CAS PubMed.
- G. Banda and I. E. Chakravarthy, Tetrahedron: Asymmetry, 2006, 17, 1684 CrossRef CAS PubMed.
- (a) R. A. Fernandes and K. Pullaiah, J. Org. Chem., 2012, 77, 9357 CrossRef CAS PubMed; (b) D. A. Chaudhari, K. Pullaiah and R. A. Fernandes, Tetrahedron: Asymmetry, 2014, 25, 1022 CrossRef CAS PubMed; (c) A. Roy, B. A. Bhat and S. D. Lepore, Org. Lett., 2015, 17, 900 CrossRef CAS PubMed.
- S. J. Gharpure, L. N. Nanda and M. K. Shukla, Eur. J. Org. Chem., 2011, 6632 CrossRef CAS PubMed.
- (a) G. Zhong, Angew. Chem., Int. Ed., 2003, 42, 4247 CrossRef CAS PubMed; (b) B. List, J. Am. Chem. Soc., 2002, 124, 5656 CrossRef CAS PubMed; (c) D. W. C. MacMillian, Nature, 2008, 455, 304 CrossRef PubMed.
- (a) P. Kumar, V. Jha and R. G. Gonnade, J. Org. Chem., 2013, 78, 11756 CrossRef CAS PubMed; (b) V. Jha and P. Kumar, RSC Adv., 2014, 4, 3238 RSC; (c) V. Jha and P. Kumar, synlett, 2014, 25, 1089 CrossRef; (d) S. V. Kauloorkar, V. Jha and P. Kumar, RSC Adv., 2013, 3, 18288 RSC; (e) S. V. Kauloorkar, V. Jha, G. Jogdand and P. Kumar, Org. Biomol. Chem., 2014, 12, 4454 RSC.
- (a) N. B. Kondekar and P. Kumar, Org. Lett., 2009, 11, 2611 CrossRef CAS PubMed; (b) P. Kumar and N. Dwivedi, Acc. Chem. Res., 2013, 46, 289 CrossRef CAS PubMed; (c) V. Jha, N. B. Kondekar and P. Kumar, Org. Lett., 2010, 12, 2762 CrossRef CAS PubMed.
- Diastereomeric and enantiomeric excess were determined using HPLC (see ESI†). In order to determine the chiral purity of (R)-ethyl-4-hydroxydecanoate 6b, it was converted into lactone 11 on treatment with p-TSA in methanol
. - The ratio of the mixture was determined by 1H-NMR of crude mixture (see ESI†).
- The protons H1 H2 H3 H4 and H5 were arbitrarily assigned to show the relative syn and anti-stereochemistry.

Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10082h |
| This journal is © The Royal Society of Chemistry 2015 |