
Advancement in methodologies for reduction of nitroarenes
Hari K. Kadam
 and
Santosh G. Tilve
*
and
Santosh G. Tilve
*
Department of Chemistry, Goa University, Taleigao Plateau, Goa-403206, India. E-mail: stilve@unigoa.ac.in
First published on 17th September 2015
Abstract
The importance of aryl amines as raw materials for various applications has spurred extensive research in developing economic processes for the reduction of nitroarenes. Developing green methodologies is now a compelling discipline for synthetic organic chemists. The recent surge in nanochemistry has led to the development of some interesting applications in nitro reduction processes. This review discusses some recent examples of reports in this field. The different methods are classified based on the source of hydrogen utilized during reduction and the mechanism involved in the reduction process.
 Hari K. Kadam | Dr Hari K. Kadam (born in Goa, India) completed his MSc (organic chemistry) with a Gold Medal in 2009 from Goa University and simultaneously passed the CSIR-UGC NET JRF exam. He completed his PhD degree in 2015 at Goa University under the supervision of Prof. S. G. Tilve. Presently, he is employed in St Xavier's College, Goa as assistant professor in chemistry. His current research interests include synthesis of bioactive heterocyclic compounds and cross coupling reactions. |
 Santosh G. Tilve | Prof. Santosh G. Tilve is a professor of organic chemistry at the Department of Chemistry, Goa University. He received his PhD degree in 1989 from Pune University under the supervision of Prof. R. S. Mali. After working in the chemical industry for six months, he started his academic career as a lecturer at Goa University. He was promoted to associate professor in 1999 and to full professor in 2007. He also worked as a visiting fellow with Prof. I. Blair at the Pennsylvania University (USA) in 2000–2002. His current research interests include asymmetric synthesis, heterocycles, green chemistry, domino reactions and nanocomposites as catalysts. |
Synthetic chemistry plays a vital role in satisfying the huge demand for organic, inorganic or biochemical materials required for various important applications. Major problems in achieving this noble task are (i) energy-expensive technologies; (ii) use of toxic solvents, reagents or catalysts; (iii) generation of harmful waste as by-products, etc.1 Most of these issues are tackled in synthetic preparations and transformations by (i) designing reactions maintaining atom economy and minimum energy usage; (ii) revealing domino processes; (iii) improving the existing processes to minimize waste; (iv) developing methods in an energy-efficient manner; (v) exploring non-toxic recyclable catalysts; (vi) replacing polluting solvent systems with aqueous medium for reactions; and (vii) using renewable sources of energy.1e,f,i,2
Aromatic amines are important intermediates in the synthesis of several nitrogen-containing biologically active compounds, agrochemicals, dyes, polymers, etc.3 They are the precursors for many synthetically important intermediates like amides, imines, azo compounds, isocyanates and diazonium salts which could be converted to various other functional groups.4 Anilines also form substructures of many pharmaceutical compounds (Fig. 1). Paracetamol5 1a, a widely used analgesic and antipyretic, is an acetyl derivative of p-aminophenol. Bicalutamide6 1b is a non-steroidal antiandrogen administered orally for the treatment of prostate cancer and hirsutism. This drug has a p-cyano-m-trifluoroaniline component in its structure. Nilutamide7 1c having a p-nitro-m-trifluoromethylaniline core in its chemical structure is an antiandrogen used in the treatment of advanced-stage prostate cancer. Erlotinib8 1d having an m-acetylenylaniline and quinazoline component is a reversible tyrosine kinase inhibitor being used in the treatment of non-small cell lung cancer and pancreatic cancer. Linezolid9 1e, a synthetic antibiotic for multi drug resistant Gram-positive bacteria, has an m-fluoro-p-morpholinoaniline component. Fosamprenavir10 1f, an anti-HIV drug and pro-drug of amprenavir, has a p-sulfonamidoaniline unit in its structure.
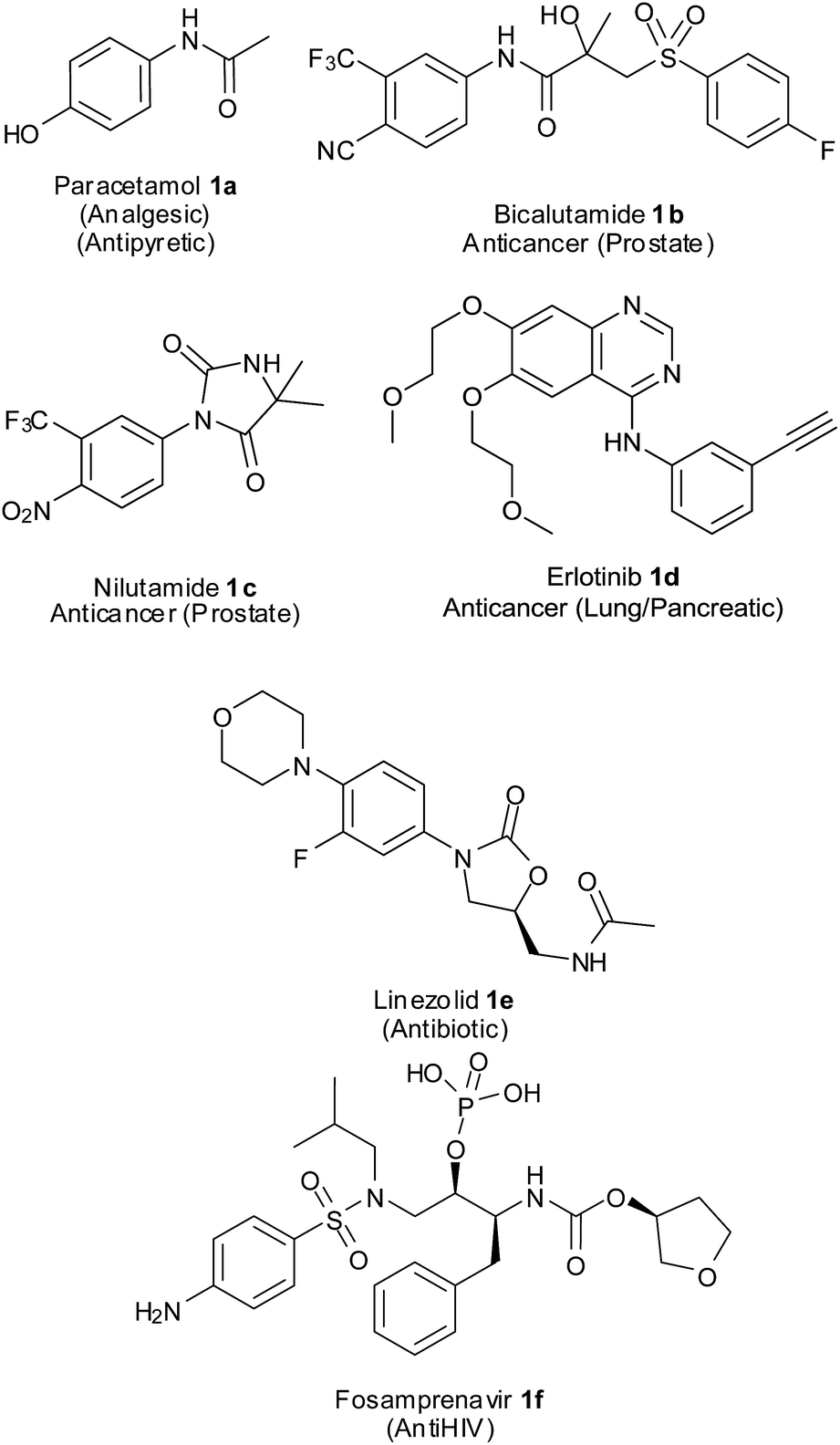 | ||
| Fig. 1 Medicinal compounds with aryl amine core structures. | ||
Reduction of nitroarenes is a most common, short and facile route employed to prepare anilines and is one of the areas where a major part of recent published work is targeted (Scheme 1).11,12 Synthetic chemists are now focusing on exploring new and efficient catalysts and developing simple and green procedures for this reaction. Selectivity in this reduction on larger scales is an important challenge in industrial processes.13 Starting with Bechamp reduction,14 a century-old process where a lot of metallic waste is generated, recent advances provide methods using catalytic metals and clean reaction conditions.
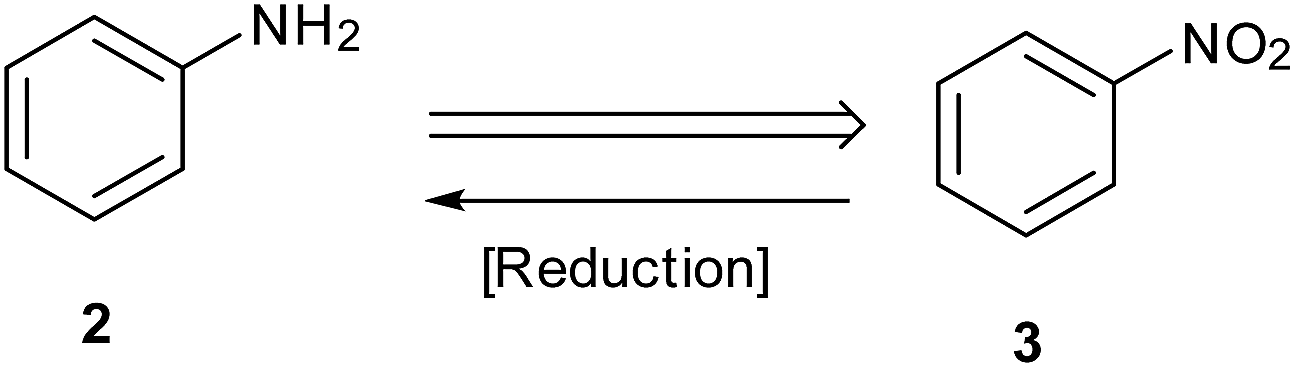 | ||
| Scheme 1 Efficient approach for synthesis of aryl amine. | ||
Our interest in achieving reduction of nitroarenes and other green methodologies15 stimulated us to compile recent progresses and achievements reported for developing facile, energy-efficient and green methodologies for reduction of nitroarenes (Scheme 2).
 | ||
| Scheme 2 Reduction of nitroarenes. | ||
The discussion is organized with respect to the use of reducing agents such as molecular H2, hydrides, hydrazine, in situ H2 generation, metal reductants, redox systems, light-induced electron transfer and biotic reduction in benign, clean, non-hazardous and non-polluting processes for reduction of nitroarenes.
(1) Hydrogen gas, (2) NaBH4, (3) silyl hydrides, (4) hydrazine hydrate, (5) in situ H2 generation, (6) direct metal, (7) MPV type redox processes using organic reducing agents (transfer hydrogenation), (8) light-induced photocatalysis, (9) biotic reduction.
1. Hydrogen gas
Molecular hydrogen in the presence of metal/metal oxide can cause clean reduction of a nitro group into an amine along with water, a benign by-product (Scheme 3). However, the formation of intermediates like N-phenylhydroxylamine (PHA), nitrosobenzene, azobenzene, azoxybenzene and hydroazobenzene and their further conversion to amines is an integral part of the reaction associated with such reductions. Metal leaching, recovery of catalyst, low catalyst loading, higher turnover cycle, use of benign solvent, low pressure conditions and compatibility of other functional groups are the problems to be addressed while designing a metal-bound catalyst. Earlier reports mostly used carbon as a support whereas newer supporting agents are now tried for efficient recovery and selectivity studies.11d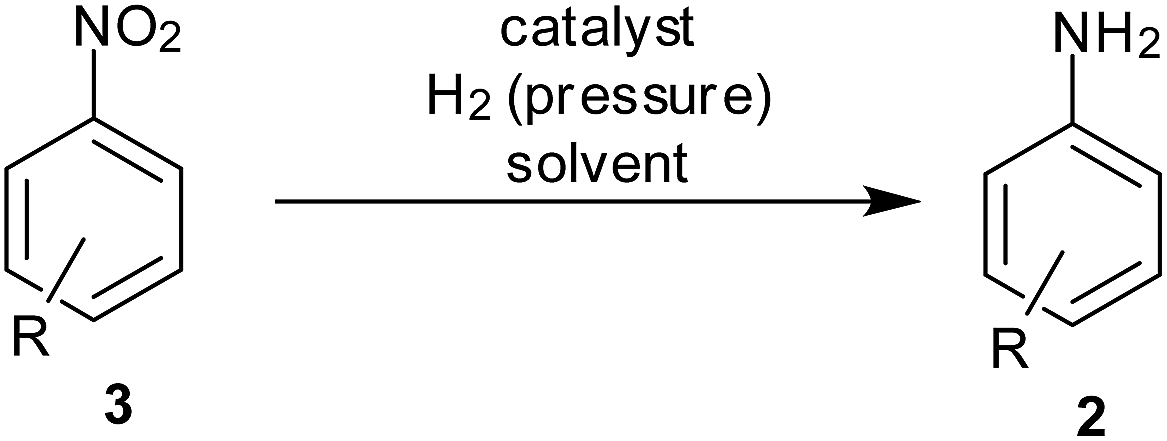 | ||
| Scheme 3 Reduction of nitroarenes using hydrogen gas. | ||
Magnetic catalysts have attracted considerable attention these days due to their ease of separation. Amine-functionalized magnetic Fe3O4 nanoparticles (NPs) supporting 1.6 mol% Pd were developed as a catalyst for reduction of aromatic nitro compounds to anilines using H2 gas in ethanol at r.t. by Ma and coworkers16 (Table 1). Here magnetic separation helped in the efficient recovery of the catalyst. Halogen (Cl, Br)- or hydroxyl group-bearing nitrobenzenes were selectively reduced to corresponding anilines with 92–99% yield. The authors report no dehalogenated products. Compared to traditional reduction (10% Pd–C), which requires 2 h for completion, the reductions took place in shorter times (1 h) with less loading of Pd due to the activating effect of the amine functionality present on the support. The reduction of 1-nitronaphthalene took a longer time (75 min). The catalyst showed excellent reusability for ten cycles with negligible leaching of the metal and TOF of 83.33 h−1. This reduction system could also reduce the double bonds in stilbene, cinnamyl alcohol and methyl cinnamate. The Heck reaction was also performed with an excellent yield using this catalyst.
| Entry | Catalysts | H2 pressure (atm) | Solvent, conditions | Ref. |
|---|---|---|---|---|
| 1 | Fe3O4–NH2–Pd | 1 | EtOH, r.t. | 16 |
| 2 | Pd/Fe3O4 | 1 | EtOH/THF, r.t. | 17 |
| 3 | Polyurea entrapped Pd nanoclusters | 1 | n-Hexanes, r.t. | 18 |
| 4 | Pd–B-mesoporous molecular sieve | 20 | scCO2, 50 °C | 19 |
| 5 | Colloidal gum acacia–Pt NPs | 1 | Water, r.t. | 20 |
| 6 | Pt/carbon nanotubes | 1 | EtOH, r.t. | 21a |
| 7 | Pt–multiwalled carbon nanotubes | 40 | Aniline, 60 °C | 21b |
| 8 | Pt–N-heterocyclic carbene NPs | 1 | THF, r.t. | 22 |
| 9 | Pt-ionic liquid | 10 | 90 °C | 23 |
| 10 | Pt–polysiloxane gel | 10 | EtOAc, r.t. – 50 °C | 24 |
| 11 | Pt/SiO2 | 1 | IPA, r.t. | 25 |
| 12 | Au–TiO2 | 9 | Toluene, 100 °C | 26a |
| 13 | Au–TiO2/Au–Fe2O3 | 9–25 | Toluene, 110 °C | 26b |
| 14 | Au-organic–inorganic hybrid SiO2 | 40 | EtOH, 100–140 °C | 27 |
| 15 | Au–ZrO2 | 10 | EtOH, 150 °C | 28 |
| 16 | Au–boronate NPs | 5 | Toluene, 100 °C | 29 |
| 17 | Pt–Au–TiO2 | 10 | EtOH, 50 °C | 30 |
| 18 | Ag–CeO2 | 6 | Dodecane, 110 °C | 31 |
| 19 | Colloidal Ni–carboxymethyl cellulose | 40 | H2O–MeOH, r.t. | 32 |
| 20 | TiO2/Ni–TiO2 | 40 | H2O–CO2, 35 °C | 33 |
| 21 | Ni–SiO2 | 20–30 | EtOH, 110 °C | 34 |
| 22 | Ru–reduced graphene oxide | 20 | EtOH–H2O, 110 °C | 35 |
| 23 | Mixed Ln–succinate–sulfate | 5 | Toluene, 90 °C | 36 |
Pd supported on magnetic Fe3O4 was developed by Amali and Rana17a for selective reduction of chloronitroarenes. These systems showed TOF of 48.5 h−1 and negligible leaching even after 10 cycles. Similarly Pd(0) was immobilized with polyethyleneimine on Fe3O4 NPs by Sun and coworkers.17b Low loading of Pd (0.25%) could also efficiently and selectively reduce 4-nitroacetophenone to 4-acetylaniline. The former was also used for Suzuki reaction and the latter for reduction of double and triple bonds as well as Suzuki–Miyaura reaction. The stability and efficient magnetic recovery of the catalyst in turn helped in enhancing the reusability up to five cycles with a slight decrease in activity. Palladium nanoclusters entrapped in polyurea prepared by Ji et al.18 exhibit dual catalytic activity for reduction of nitro compounds and dehalogenation of aromatic chlorides in atmospheric hydrogen with 100% yield for reduction of nitro compounds at room temperature and >99% yield for dehalogenation of aromatic chlorides under refluxing methanol conditions. This immobilizing method was particularly effective and eliminated the need of special chelating groups. However, the authors have not addressed the usual selectivity study for problems of concurrent dehalogenation.
Supercritical CO2 as a green solvent was used along with Pd NPs supported on B–MCM-41 as catalyst and H2 gas for hydrogenation of nitro aromatics by Chatterjee et al.19 The o-, m- and p-chloronitrobenzenes (CNB) were reduced to the corresponding amines with high selectivity of >99% and conversion in the order of p > m > o. This system could also reduce a nitrile group and a phenol to cyclohexanone. However, in all the above cases selectivity in the reduction between nitro and functional groups like olefin, aldehyde, cyano and benzyl ether was not studied.
Hydrogenation of nitroarenes catalyzed by gum acacia-supported Pt colloids with 0.24 mol% catalyst loading in water at r.t. using H2 at 1 atm is described by Sreedhar et al.20 This catalytic condition was inert to halogens, aldehydes and ketones with selective reduction of nitro group in 68 to 95% yield. The yields were found to be consistent for 5 cycles with no leaching of metal.
Newer carbon supports have attracted the attention of people working in the field of catalysis. Pt supported on carbon nanotubes (Pt/CNT) and PtM/CNT (M = Mn, Fe, Co, Ni and Cu) were studied as catalysts for selective hydrogenation of m- and o-CNB to corresponding chloroanilines by Han and Li.21a All metals studied except Cu were found to enhance the catalytic behavior of Pt/CNT and PtFe/CNT was found to be the best. Solvent-free selective hydrogenation of nitrobenzene to aniline using ultrafine Pt deposited on carbon nanotubes is reported by Sun et al.21b High turnover frequency (69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 h−1) without accumulation of PHA is noteworthy in this process. However, reusability, metal leaching and selectivity to other functional groups were not studied for this process.
900 h−1) without accumulation of PHA is noteworthy in this process. However, reusability, metal leaching and selectivity to other functional groups were not studied for this process.
Chaudret et al. have described the chemoselective reduction of a series of functionalized nitroarenes with H2 gas (1 bar) at r.t. using Pt NPs stabilized with N-heterocyclic ligands as catalyst.22 Sensitive functionalities such as carbonyl, olefins as well as halogens were tolerated in this reduction. Ethyl-4-nitrocinnamate was reduced with 79% selectivity, 4-chloronitrobenzene with 95%, 4-nitrobenzaldehyde with 94% and 3-nitropyridine with >99%.
Ionic liquids (IL) are pursued as green alternatives for toxic volatile solvents. IL-like copolymer stabilized Pt nanocatalysts were studied for selective hydrogenation of 2,4-dichloro-3-nitrophenol to 2,4-dichloro-3-aminophenol using H2 gas in different IL by Yuan et al.23 The IL system containing an alcohol group displayed better selectivity, recyclability (9 times) and higher turnover number (2075).
Polysiloxane gels containing Pt species, [Pt]@SiC6 and [Pt]@SiC6-TAA, were demonstrated by Nagashima and coworkers as recyclable heterogeneous catalysts for reduction of various nitro compounds to their corresponding amines with other functional groups (ester, ketone, benzyl ether, benzyl alcohol, amide and chloro) remaining intact.24 Turnover number up to 10000 was achieved, the catalyst recovery/reuse was done five times and metal leaching was beyond detection limits. Aliphatic nitro groups were reduced rather slowly at room temperature.
Substituted nitro aromatics were selectively hydrogenated to the corresponding N-aryl hydroxylamines in excellent yields (up to 99%) using supported platinum catalysts such as Pt/SiO2 under a hydrogen atmosphere (1 bar) at room temperature by Takenaka et al.25 This reduction was carried out in IPA with DMSO and n-BuNH2 as additives.
The chemical community had ignored gold due to its low reactivity, but recently its unique catalytic properties have drawn the attention of numerous research groups, which has been reflected in a number of research publications in the literature.
Chemoselective reduction of nitroarenes containing double bonds, carbonyl, nitrile or amide groups on supported gold NPs (Au/TiO2 and Au/Fe2O3), using a batch reactor under H2 pressure, was demonstrated by Corma et al.26a This group also used Au on TiO2 as a hydrogenation catalyst to prepare azo compounds directly from nitroaromatics through a two-step (hydrogenation followed by aerobic oxidation), one-pot, one-catalyst reaction.26b
Highly dispersed gold NPs supported on organic–inorganic hybrid silica were shown to exhibit good catalytic activity and stability for liquid phase catalytic hydrogenation of aromatic nitro compounds by Tan et al.27 p-CNB was reduced with 80% selectivity with a significant amount of p-chloronitroso intermediate remaining. Similarly hydrogenation of CNBs to chloroanilines with complete selectivity was reported over Au/ZrO2 catalyst with H2 gas in ethanol by He et al.28 Recently, gold NPs embedded in boronate self-assemblies were used for selective reduction of 4-nitrostyrene using H2 gas.29 Adding small amounts of Pt entities (0.01–0.03 wt%) onto the Au surface of a Au/TiO2 catalyst was shown to be an efficient approach to improve the catalytic activity of Au for the hydrogenation of p-CNB by He et al.,30 where the C–Cl bond remained intact. Excess amounts of Pt (>0.03 wt%) and high reaction temperatures caused the occurrence of the undesired catalytic hydrodechlorination reaction of p-CNB. Reusability of this catalyst system was demonstrated for five cycles without leaching of any of the metals.
Ag@CeO2 core–shell nanocomposite was used as a catalyst for reduction of nitro compounds to anilines with H2 gas by Kaneda and coworkers.31 This catalyst helped to achieve complete selectivity towards nitro reduction in the presence of double bonds with >95% yield. Selectivity in the presence of other functional groups particularly like halogen or aldehyde would have been interesting but appears not to have been studied. This system could also reduce oxiranes to alkenes.
Among the coinage metals, Ni is preferred over other metals because of its low cost. A biopolymer-inorganic catalyst system involving colloidal Ni and carboxymethylcellulose was reported for reduction of nitroaromatics using H2 gas at r.t. in MeOH–water mixture by Ali and coworkers.32 Various aniline products were obtained with a substrate:catalyst ratio of 100:1 and 40 bar H2 gas in 79–96% yields. Reduction was achieved in the presence of ester, OH and NH2 groups on the aromatic ring. This system was also useful for reduction of ketone to secondary alcohol. Low pressure CO2–water system with Ni was applied for reduction of nitrobenzene to aniline by Arai and coworkers.33 Ni was supported on Al2O3 for this reduction. Similarly CNB was reduced to chloroaniline with Ni/TiO2 in low pressure CO2 (3 MPa)–water system by the same group. Zheng et al.34a have described Ni/SiO2 catalyst for selective reduction of nitroarenes to anilines using H2 gas wherein ketone, aldehyde, chloro and amide functionalities were found to be unaffected. Magnetic recovery and reusability of this supported catalyst were also demonstrated for five cycles. Jiang's group has recently presented a one-pot synthesis of Ni–Ni–Fe2O4/carbon nanofiber composites from biomass and utilized them as a catalyst for selective hydrogenation of aromatic nitro compounds with hydrogen gas.34b
Graphene and graphene oxide materials are studied for various applications in material science and this trend has been followed even in catalysis because of their applications as supports and also their ability to enhance the property exhibited by a catalyst.
A reduced graphene oxide (RGO)-supported ruthenium (Ru) catalyst was prepared by Wang et al. and applied for the selective hydrogenation of p-CNB to p-chloroaniline, exhibiting a turnover frequency (TOF) of 1800 h−1 and a selectivity of 99.6% at complete conversion of p-CNB. Ketone functionality was also well tolerated during the reduction.35 Here Ru NPs were in an electron-deficient state due to the electron transfer between the NPs and the RGO sheets. No loss in efficiency of this catalyst system was observed for ten runs with minimal leaching of Ru (0.2%).
Mixed lanthanide succinate–sulfate isostructural 3D polymeric metal–organic frameworks of monoclinic space group have also been used for reduction of the nitro group by Monge and coworkers.36 Other reducible groups like aldehyde, cyano, halogen (Br, I) remained unaffected during the reduction.
Though catalytic hydrogenation is routinely employed in industry and in research laboratories, it has the distinct disadvantage of the requirement of special equipment to handle high-pressure and inflammable H2. Also a large amount of hydrogen is wasted and is usually lost to the atmosphere after the reaction is over.
2. NaBH4
In situ generation of hydrogen during the reduction process can avoid the use of sophisticated equipment required for handling hydrogen gas and wastage due to excess use of gas under pressure. NaBH4 has been employed as a clean source of hydrogen generation in fuel cells using different metal-bound catalysts. The same system could also be used for reduction of nitro to amine functionality with formation of non-toxic sodium borate as a by-product (Scheme 4).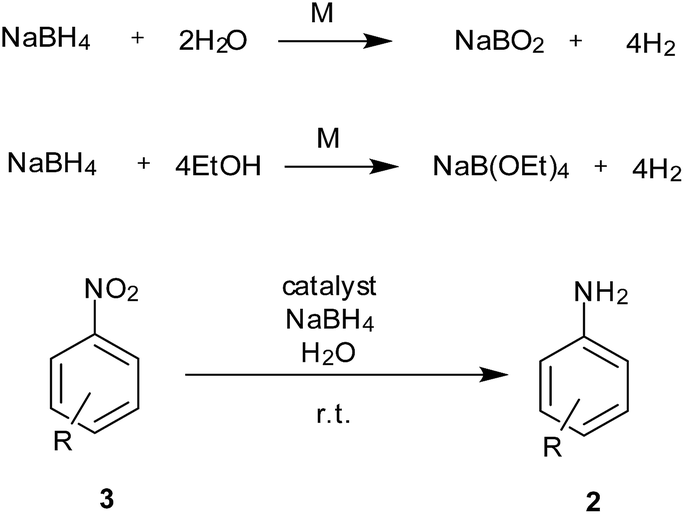 | ||
| Scheme 4 Reduction of nitroarenes using NaBH4 (3–5 equiv.). | ||
Ease of handling NaBH4 is also an added advantage in this process. The functional groups which get reduced with NaBH4 are usually not tolerated during usage of this reduction process.
Synthesis of Pd-incorporated poly(3,4-ethylenedioxythiophene) (PEDOT) matrix in aqueous medium was achieved and its catalytic activity was demonstrated using a model reaction, i.e. reduction of 4-nitrophenol to 4-aminophenol using NaBH4, by Harish et al.37a (Table 2). Similar solid-supported Pd(0)-catalyzed highly chemoselective reduction of nitroarenes to the corresponding anilines was accomplished in MeOH–H2O mixture by Shil et al.37b This catalyst showed high compatibility with various reducing agents like NaBH4, Et3SiH, and NH2NH2·H2O, and a large number of reducible functional groups such as sulfonamide, amide, carboxylic acid, ester, alcohol, halide, heterocycle, nitrile, alkene, carbonyl, O-benzyl, and N-benzyl were tolerated.
| Entry | Catalyst | Ref. |
|---|---|---|
| a EtOH, 100 °C.b NH3BH3, EtOH.c EtOH.d MeOH. | ||
| 1 | Pd–poly(3,4-ethylenedioxythiophene) matrix | 37 |
| 2 | Pd(II) phthalocyaninea | 38 |
| 3 | Au–Fe3O4 nanocatalyst | 39a |
| 4 | Cu(II)silica@Fe3O4 composites | 39b |
| 5 | Au-Nano Active MgO Plus | 40 |
| 6 | Au nanorods, KBH4 | 41 |
| 7 | Au–epigallocatechin-3-gallate–collagen fiber | 42 |
| 8 | Au–resorcinarene NPs | 43 |
| 9 | Au–alumina/membrane | 44 |
| 10 | Au–boronate NPs | 30 |
| 11 | Au-double hydrophilic block copolymer | 45 |
| 12 | Au–graphene hydrogel | 46 |
| 13 | Au–TiO2b | 47 |
| 14 | Ag–halloysite nanotubes | 48 |
| 15 | Ag quantum clusters | 49 |
| 16 | Ag–Au–Fe3O4–carbon composite | 50 |
| 17 | Ag–graphite–polyamidoamine dendrimer | 51 |
| 18 | Hollow Ag nanospheres | 52 |
| 19 | (Pt/Au) NPs | 53 |
| 20 | CuBr2c | 15b |
| 21 | Cu NPs | 54a |
| 22 | Cu–ferrite graphene hybrid | 54b |
| 23 | Pd/Cu/graphene | 54c |
| 24 | Cu/MIL-101(Cr) nanocomposites | 55 |
| 25 | Co3S4 | 56 |
| 26 | Co–Co2Bd | 15c |
Pd(II) phthalocyanine was also used with low catalyst loading up to 1 mol% along with NaBH4 in EtOH by Verma et al.38
Dumbbell- and flower-like Au–Fe3O4 heterostructures have been fabricated by thermal decomposition of an iron oleate complex in the presence of Au NPs using different sizes of Au NPs as the seeds and employed as magnetically recyclable catalysts (for p-nitrophenol and 2,4-dinitrophenol reduction) by Lin and Doong.39a Similarly, Cu(II) NPs on silica Fe3O4 support were used for reduction of nitroarenes with NaBH4 in aqueous medium at r.t. by Sharma et al.39b Other reducible moieties like CN and halides were retained during reduction.
Nanocrystalline magnesium oxide-supported gold NPs were used as a recyclable heterogeneous catalyst for reduction of nitroarenes to anilines using sodium borohydride in double distilled water at room temperature by Maheswaran and coworkers.40 This reduction system could tolerate various substitutions of the aromatic ring, like F, Cl, Br, I, OCH3, COOMe, vinyl, CN, OH and NH2. After the reduction the spent catalyst could be recycled by centrifugation and reused. There was a slight loss in recovery, which resulted in a marginal decrease in efficiency.
Uniform-sized gold nanorods have been prepared by Bai et al.41 via a three-step seed-mediated growth method using a long-chain ionic liquid (IL, C12mimBr) as a capping agent and exhibited excellent catalytic efficiency for the reduction of p-nitrophenol and p-nitroaniline. Size-controlled Au NPs supported on collagen fiber (CF) were prepared by Shi and coworkers.42 Epigallocatechin-3-gallate, a typical plant polyphenol, was grafted onto CF surfaces to serve as a reducing/stabilizing agent, so that the Au NPs were generated on the CF surface without introduction of extra chemical reagents or physical treatments. These stabilized Au NPs were found to be active heterogeneous catalysts for the reduction of 4-nitrophenol to 4-aminophenol in aqueous phase. The catalyst was recovered simply by filtering and successfully used for 20 cycles with conversion of >98%.
Resorcinarene-functionalized Au NPs were prepared in aqueous solution in the presence of amphiphilic tetramethoxyresorcinarene tetraaminoamide by Yan and coworkers.43 The catalytic activity of the obtained Au NPs for the reduction of aromatic nitro compounds was investigated. Layer-by-layer deposition of polyelectrolyte/Au NP films in porous alumina, track-etched polycarbonate and nylon substrates gave catalytic membranes that showed high catalytic activity in the selective reduction (98%) of p-nitroaromatic compounds containing cyano, chloro, and vinyl with sodium borohydride, as described by Bruening and coworkers.44 The reduction of nitrocyclohexane was incomplete, giving corresponding nitroso (73%) and amine (27%) compounds.
4-Nitrophenol was reduced with NaBH4 using Au–boronate NPs.29 Water-dispersible Au NPs using a double hydrophilic block copolymer, poly(ethylene oxide)-block-poly(acrylic acid), as a template were prepared and found to be highly effective in catalyzing the reduction of a series of nitroarenes by Kim and coworkers.45 However, selectivity studies were not performed with this catalyst system.
A cylindrical piece of Au/graphene hydrogel, 1.08 cm in diameter and 1.28 cm in height, was synthesized through the self-assembly of Au/graphene sheets under hydrothermal conditions by Li et al.46 The hydrogel, containing 2.26 wt% Au, 6.94 wt% graphene, and 90.8 wt% water, exhibited excellent catalytic performance towards the reduction of 4-nitrophenol to 4-aminophenol. The high catalytic activity arises from the synergistic effect of graphene: (1) the high adsorption ability of graphene towards 4-nitrophenol, providing a high concentration of 4-nitrophenol near to the Au NPs on graphene; and (2) electron transfer from graphene to Au NPs, facilitating the uptake of electrons by 4-nitrophenol molecules.
Quantitative reduction of nitroarenes into anilines and nitroalkanes into alkylhydroxylamines by an ammonia borane complex was achieved using a catalyst of gold NPs supported on titania, even at a ppm loading level, by Stratakis and coworkers.47 Reducible functional groups like benzyl ether, halogen (Cl, Br), ester and nitrile groups remained intact while aldehyde and keto groups got reduced. In the case of 3-nitrostyrene, 10% over-reduction of the double bond was observed. Inert atmosphere is required for this reduction process to avoid formation of minor amounts of azoxyarenes. The authors have proposed a mechanism based on evidence that amines are obtained from hydroxylamines without intervention of nitrosobenzene via gold hydride species.
Silver NPs supported on halloysite nanotubes (Ag/HNTs), with Ag content of about 11%, were used for the catalyzed reduction of 4-nitrophenol with NaBH4 in alkaline aqueous solutions by Liu and Zhao.48 Quantum clusters of silver such as Ag7(H2MSA)7 and Ag8(H2MSA)8 (H2MSA, mercaptosuccinic acid) were synthesized by the interfacial etching of Ag NP precursors and were loaded on metal oxide supports to prepare active catalysts such as Al2O3@Ag7,8, SiO2@Ag7,8, TiO2@Ag7,8, and Fe2O3@Ag7,8 by Pradeep and coworkers.49 These catalysts showed enhanced catalytic activity for the reduction of nitrophenols to aminophenols.
Heterostructure Ag–Au bimetallic nanocrystals supported on Fe3O4@carbon composite microspheres were synthesized by a facile and controllable approach by Guo and coworkers,50 wherein the Ag nanocrystals attached on the Fe3O4@carbon microspheres were prepared first and served as reductant for the galvanic replacement reaction with the Au precursor (HAuCl4). They could give high yields for reduction of substituted nitroaromatic compounds, irrespective of linked electron-donating or electron-withdrawing groups.
Hyperbranched polyamidoamine (PAMAM) dendrimers were grafted on a graphite surface and Ag NPs were synthesized within the graphite-grafted PAMAM dendrimer templates and applied as a nanocatalyst for the reduction of nitroaromatics by Rajesh and Venkatesan.51 The efficiency of this system has been demonstrated through the reduction of halonitroarenes without dehalogenation in the halogeno-substituted nitrobenzenes and selective reduction of nitro groups in the presence of imine functionality under mild conditions.
Hollow silver nanosphere colloids were prepared by a simple reaction of silver nitrate (AgNO3), sodium hydroxide (NaOH) and hydroxylammonium hydrosulfate ((NH2OH)2·H2SO4) in the presence of gelatin by Parikh and coworkers.52 Superior catalytic performance was observed in 4-nitrophenol to 4-aminoaniline reduction in the presence of freshly prepared ice cold aqueous solution of sodium borohydride at room temperature.
Catalytic reduction of 4-nitrophenol by sodium borohydride was achieved by Ballauff and coworkers in the presence of Pt/Au NPs embedded in spherical polyelectrolyte brushes, which consist of a polystyrene core onto which a dense layer of cationic polyelectrolyte brushes are grafted. The average size of these NPs was approx. 2 nm.53
We have reported copper(II)bromide as a procatalyst for the in situ preparation of active Cu NPs for the efficient reduction of nitroarenes using sodium borohydride.15b Acid, chloro, hydroxyl, benzyl ether and amino functionalities remained intact while olefin and cyano were affected.
Gradzielski et al.54a synthesized copper NPs using poly(acrylic acid) and utilized them for catalytic reduction of 4-nitrophenol to 4-aminophenol. The activity was found to increase as the particle size decreased. Superparamagnetic Cu–ferrite–graphene hybrid nanocomposites were used for reduction of nitroarenes by Wang and coworkers.54b The ferrite component helped in efficient recovery without loss in catalytic activity. Pd–Cu NPs supported on graphene were used by Feng et al. for chemoselective reduction of nitroarenes with NaBH4 in the presence of CN, ester, halogens, etc.54c Cu nanostructures of various shapes and sizes such as nanospheres, nanowires and nanorods were synthesized by Kaur and Pal and their catalytic activities were studied for nitroaromatic reduction.54d Cu nanowires (length ≈ 4–6 μm and width ≈ 60–80 nm) were found to exhibit superior catalytic activity. Similarly 3-nitro-4-methoxyacetylaniline was selectively reduced by Feng et al. to 3-amino-4-methoxyacetylaniline using Cu NPs as catalyst and NaBH4 as hydrogen source in water.54e
Wu et al.55 loaded Cu NPs on a MIL-101(Cr) metal–organic framework which showed enhanced catalytic activity for the reduction of aromatic nitro compounds.
Cobalt sulfide, Co3S4, was recently reported for such a reduction using NaBH4 in EtOH under sonication.56 The halogens were unaffected during this nitroreduction.
Magnetically recoverable and recyclable Co–Co2B nanocomposites are described for the catalytic and chemoselective reduction of nitroarenes using sodium borohydride from our laboratory.15c Halogen, benzyl ether and acid functionalities remained undisturbed while cyano and aldehyde groups got reduced.
Though NaBH4-mediated reductions are safer to handle compared to catalytic hydrogenations, they have the problem of workup to extract the product from the aqueous reaction medium. Also excess of NaBH4 is required to complete the reduction process. Further, metal reacting with NaBH4 generates hydrogen, which needs to be taken care of when large-scale reductions are to be carried out. Also in most of the above cases the selectivity problem was not addressed; rather, the work centered on making NPs and demonstrating the usefulness of the NPs for catalytic processes.
3. Silyl hydrides
Nitro reduction with silyl hydrides proceeds through the nitroso and hydroxylamines route; the exact mechanism for this reduction process is not clear. It may take place via metal-catalyzed hydrosilylation or via hydrogenation with evolved hydrogen gas (Scheme 5). | ||
| Scheme 5 Reduction of nitroarenes using silyl reagents. | ||
As early as 1973, Lipowitz and Bowman reported the first example of polymethylhydrosiloxane (PMHS)-mediated Pd/C-catalyzed reduction of nitrobenzene to aniline57a (Table 3). A combination of Pd(OAc)2, aq. KF and PMHS was reported for reduction of aromatic nitro groups to amines at room temperature in high yields with wide functional group tolerance and short reaction times by Rahaim and Maleczka.57b Steric hindrance by one ortho substituent did not slow the reduction process while two ortho substituents did. Electron-donating groups were well tolerated except for 4-nitrothioanisole (10% yield) while 2-nitrothiophene was reduced successfully. Electron-withdrawing groups like acid, ester, amide, benzylic ketone, fluoro and trifluoro were unaffected. The presence of a nitrile group (o, m) led to longer times being required to form the corresponding aminonitriles while reduction of p-nitrobenzonitrile stopped at p-(hydroxyamino)benzonitrile. Selectivity in the reduction of 4-nitrobenzaldehyde was 73% and in the reduction of 1,4-dinitrobezene was 72%. Interestingly TBS-protected nitrophenol was reduced selectively (93%) in spite of the presence of KF. However, complete dehalogenation was observed for chlorobromonitrobenzenes while aliphatic bromide remained intact. Olefin remained unaffected during the course of the reaction. Methyl-5-nitro-2-furanoate was reduced successfully whereas the reduction of 5-nitrobenzamidazole required modification of the protocol sequence. Aliphatic nitro groups (primary and secondary) were reduced to hydroxylamines with the same system by replacing PMHS/KF with triethylsilane.
| Entry | Catalyst | Silanes (equiv.) | Solvent/conditions | Ref. |
|---|---|---|---|---|
| 1 | Pd(OAc)2 | PMHS (4)/KF | THF/H2O, r.t. | 57 |
| 2 | ReOCl3(PPh3)2 | PhMe2SiH (36) | Toluene, 110 °C | 58 |
| 3 | Fe(acac)3 | TMDS (4) | THF, 60 °C | 59 |
| 4 | FeBr2, PPh3 | PhSiH3 (2.5) | Toluene, 110 °C | 60 |
| 5 | Fe(II) phthalocyanine | Ph2SiH2 (3) | EtOH, 100 °C | 38 |
| 6 | Au–Fe3O4 | TMDS (4–10) | EtOH, r.t. | 61 |
Alternatively, the reduction of aromatic nitro compounds to the corresponding amines with silanes catalyzed by high-valent oxo-rhenium complexes is reported by Fernandes and coworkers.58a The catalytic systems PhMe2SiH/ReIO2(PPh3)2 (5 mol%) and PhMe2SiH/ReOCl3(PPh3)2 (5 mol%) reduced efficiently a series of aromatic nitro compounds in the presence of a wide range of functional groups such as ester, halogen, amide, sulfone, lactone, and benzyl. This methodology also allowed the regioselective reduction of dinitrobenzenes to the corresponding nitroanilines and the reduction of an aromatic nitro group in the presence of an aliphatic nitro group. Similarly, Wilkinson's catalyst, RhCl(PPh3)3, was also used along with Et3SiH in refluxing toluene for reduction of nitroarenes.58b
1,1,3,3-Tetramethyldisiloxane (TMDS)/Fe(acac)3 was used for nitro reduction and the product amines were isolated as hydrochloride salts with good to excellent yields by Lemaire and coworkers.59 Nitrile, acid, ester and bromo groups were well tolerated while p-nitrobenzaldehyde gave p-hydroxymethylaniline. m-Dinitrobenzene was selectively reduced to m-nitroaniline.
An iron-based catalytic system consisting of FeBr2–Ph3P was used for the reduction of nitroarenes with organosilanes by Beller's group.60 Except for fluorine substituent, high yields of anilines were obtained for halonitrobenzenes without a significant amount of dehalogenated products. The catalyst also showed selectivity with challenging substrates with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C
O, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, CC, and NO2 groups. However, hydrosilylation failed in the case of 3-nitrostyrene and 4-nitrophenylacetate.
N, CC, and NO2 groups. However, hydrosilylation failed in the case of 3-nitrostyrene and 4-nitrophenylacetate.
Fe(II) phthalocyanines were also used with diphenylsilane as hydrogen source in refluxing ethanol.38 This method was applied on a gram scale for the conversion of p-nitrotoluene to p-toluidine.
A magnetically separable gold-nanoparticle catalyst was prepared and it showed excellent activity for chemoselective reduction of nitroarenes with hydrosilanes.61 Selective reduction of 4-fluoro- and 4-chloronitrobenzene required only 1 mol% Au while 40 mol% was required for complete reduction of 4-bromonitrobenzene, and 4-iodonitrobenzene could not be reduced. Ketone, ester, amide, cyano, alkene, benzyloxy and carbobenzyloxy functional groups survived during nitro reduction. The activity of the catalyst reduced during its reuse but increasing the quantity of reducing agent could compensate for this reduced activity.
Again like NaBH4-mediated reductions, the problem of work up, scaling up and use of excess reducing agent cannot be avoided for this system. However, the selectivity in the reduction process looks to be promising and further developments are expected.
4. Hydrazine hydrate
Hydrazine hydrate is known to decompose in the presence of trace amounts of transition metal to hydrogen and benign N2 gas. The in situ generation of hydrogen gas on an active metal surface thus facilitates the reduction process.The hydrogen release from this reaction can be used for reduction processes while avoiding elaborate hydrogenation apparatus required for hydrogen gas (Scheme 6). However, the toxicity and its well-known use as rocket fuel may have to be taken into account during large-scale reduction processes.
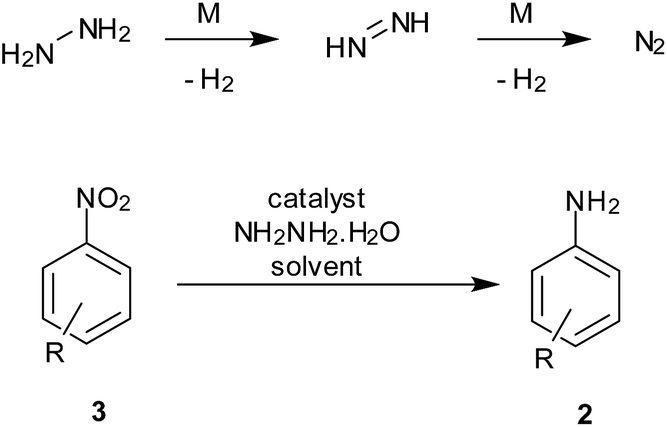 | ||
| Scheme 6 Reduction of nitroarenes using hydrazine (1.2–10 equiv.). | ||
Polymeric PEG-35k–Pd NPs were used by Yadav et al.62 for reduction of nitro compounds to amines with hydrazine hydrate as a reducing agent at 90 °C (Table 4). This reduction method was inert to halogens giving haloanilines in quantitative yield. The catalyst was recycled by centrifugation and could be used for a maximum of 8 cycles.
| Entry | Catalyst | Solvent/conditions | Ref. |
|---|---|---|---|
| 1 | PEG-35k–Pd NPs | 90 °C | 62 |
| 2 | Pd–C nanospheres | EtOH:H2O, 80 °C |
63 |
| 3 | Fe3O4 NPs | EtOH, 80 °C | 64a |
| 4 | Fe(acac)3 | MW, 150 °C | 64b |
| 5 | Graphene–Fe3O4 | 70 °C | 65 |
| 6 | Iron oxide hydroxide, polymer-supported NH2NH2 | iPrOH, 80 °C | 66 |
| 7 | FeSO4–Fe phthalocyanine | EtOH:H2O, 120 °C |
67 |
| 8 | Rh–Fe3O4 | EtOH, 80 °C | 68 |
| 9 | Hollow Rh nanocomposite | EtOH, 80 °C | 69 |
| 10 | Rh-porous ionic copolymer | EtOH, 60 °C | 70 |
| 11 | Zn phthalocyanine | PEG-400, 100 °C | 71 |
| 12 | Zn or Mg, hydrazine glyoxalate | r.t. | 72 |
| 13 | Co–Co2B | MeOH, r.t. | 15c |
| 14 | (Bu4N)[Ni(toluene-3,4-dithioalate)2] | THF, reflux | 73 |
| 15 | MoS2 | Toluene, 60–80 °C | 74 |
| 16 | PVP-stabilized Ni or Co | H2O, r.t. | 75a |
| 17 | Co–Mo2C/activated carbon | Reflux | 75b |
| 18 | Carbon/graphite | iPrOH, reflux | 75c |
| 19 | Multiwalled carbon nanotubes | EtOH, 100 °C | 76a |
| 20 | Boron–pyrolytic graphene oxide | 90 °C | 76b |
Palladium NPs immobilized on carbon nanospheres are reported to catalyze reduction of nitroaromatic compounds with 1.36% Pd using hydrazine hydrate in ethanol–water mixture as described by Yu et al.63a The authors claim that they could selectively reduce the nitro group in the presence of other reducible vinyl and aldehyde groups. It may be noted that no hydrazone product or the alcohol product is reported though hydrazine was used in excess (1:10 molar ratio). The low Pd loading (1.36%) also helped to retain halogen in the reduced products. In the case of 3-nitrophenol and 4-methylnitrobenzene, the main byproducts were corresponding azo and azoxy intermediates. Similarly Pd/C was also studied for selective reduction of halogenated nitrobenzenes using hydrazine hydrate under reflux or microwave (MW) conditions by Li and coworkers.63b
Readily available and magnetically separable Fe3O4 NPs were utilized for recyclable and efficient nitroarene reduction.64a Reducible functional groups like halogen, ester, benzyloxy ether, amide and benzyl alcohol remained intact while only 45% selectivity was obtained for the reduction of ethyl 4-nitrocinnamate. Aliphatic nitro compounds were reduced less efficiently.
In situ generated iron oxide nanocrystals were used for reduction of nitroarenes using MW irradiation by Kappe's group.64b This method using hydrazine hydrate as reducing agent yielded anilines in quantitative yields without affecting halogens, esters, amides or nitriles. Reusability studies suggested that it is effective for 3 cycles. The authors also demonstrated that the reduction could be carried out in a continuous flow method on an industrial scale. After the reduction process the colloidal Fe3O4 nanocrystals agglomerate and can be selectively removed by using a simple magnet.
Graphene–Fe3O4 nanocomposite (G–Fe3O4) and superparamagnetic G–Fe3O4 were synthesized by a chemical co-precipitation method and used as an efficient catalyst for the reduction of nitroarenes with hydrazine hydrate by Zhang et al., Wang and coworkers and Shokouhimehr et al.65 4-CNB was selectively reduced without any dehalogenation. The catalytic activity did not decrease to any extent in the five cycles studied.
Iron oxide hydroxide catalyst was used for reduction of nitroarenes to anilines with polymer (D113, macroporous weak acidic ion-exchange resin)-supported hydrazine hydrate in refluxing isopropanol by Shi and coworkers.66 Anilines were obtained in 93 to 99% yields without chlorides and esters being affected.
Iron phthalocyanine and iron sulfate-catalyzed reduction of nitroarenes to anilines was reported with hydrazine hydrate as hydrogen source in a mixture of water and ethanol by Sharma et al.67 This method was applied on a gram scale to substrates with substituents like acid, nitrile, sulfonamide, hydroxyl, O-benzyl, N-benzyl, lactones, etc. 4-Chloro-2-nitrophenol was selectively reduced to the corresponding aniline without affecting other functionalities. Also other heterocyclic nitro compounds like nitroisoquinoline, nitroindole, nitrothioindole, etc. were successfully reduced to corresponding amines.
Rh–Fe3O4 heterodimer nanocrystals were prepared by controlled one-pot thermolysis. The nanocrystals exhibited excellent activities for the selective reduction of nitroarenes and alkenes as reported by Hyeon et al.68 Hollow Rh nanocomposites also showed similar results.69 A highly active and selective Rh/highly porous ionic copolymer nanocatalyst for the reduction of nitroarenes into corresponding anilines with hydrazine monohydrate under mild conditions was also reported by Luo et al.70 The halonitrobenzenes were reduced successfully without any dehalogenated products.
Zn phthalocyanine was used as a catalyst (1 mol%) for reduction of aromatic nitro compounds to anilines using N2H2·H2O as reducing agent and PEG-400 as solvent by Singh and coworkers.71 Various functional groups like acid, ester, amide, sulfonamide, cyano, halogen, benzyloxy and benzyl amine were unaffected in this nitro reduction. 3-Nitrostyrene and 1,3- and 1,4-dinitrobenzenes were reduced selectively while 1,2-dinitrobenzene showed moderate conversion (58%). Exclusive formation of benzotriazole was obtained when excess hydrazine hydrate was used during reduction of 1,2-dinitrobenzene. Aromatic nitro compounds were selectively and rapidly reduced at r.t. to corresponding amines in good yields by employing hydrazine glyoxalate in the presence of Zn or Mg powder by Raju et al.72 Halonitrobenzenes were reduced to corresponding amino benzenes without dehalogenation. p-Nitrocinnamic acid is reduced to p-aminocinnamic acid with no reduction of the olefin bond. No selectivity is reported for dinitrobenzene as diaminobenzene formation was observed.
Magnetically recoverable and recyclable Co–Co2B nanocomposites described earlier from our laboratory for the catalytic and chemoselective reduction of nitroarenes using sodium borohydride have also been demonstrated for reduction of nitro groups using hydrazine hydrate.15c Halogen, ester, benzyloxy, nitrile and aliphatic nitro functionalities remained intact while allyloxy group showed 92% selectivity.
Transfer hydrogenation of aromatic nitro compounds by hydrazine to the corresponding anilines is catalyzed by (Bu4N)[Ni(toluene-3,4-dithioalate)2] in refluxing THF.73 Nitroarenes with electron-withdrawing groups are more easily reduced than those with electron-donating groups. In most cases anilines are the sole products while, in a few cases, N-phenylhydroxylamines are formed as intermediates and chief products at lower catalyst loading or for shorter reaction times.
Commercial MoS2 was found to be a highly selective catalyst for the reduction of nitrobenzenes to the corresponding anilines with hydrazine under mild conditions by Huang et al.74 Very high selectivity is observed in the reduction of halonitrobenzenes and styrylnitro compounds. Polyvinylpyrrolidine-stabilized Ni or Co NPs were used for selective reduction of nitroarenes in the presence of Cl, Br, I, CN. Aliphatic nitro compounds were also reduced using this system.75a Quantitative conversion of nitroarenes to anilines was obtained with cobalt-modified Mo carbide supported on activated carbon in refluxing hydrazine hydrate. Sensitive reducible groups like Cl, ester, and aldehyde were tolerated during reduction.75b Reduction of nitroaromatics to anilines by hydrazine was also studied using carbon or graphite as catalysts.75c
Multiwalled carbon nanotubes were functionalized with small organic molecules containing specific ketonic carbonyl groups through noncovalent van der Waals and π–π interactions and utilized as metal-free catalysts for reduction of nitroarenes.76a Boron-doped pyrolytic graphene oxide was synthesized and explored for efficient reduction of nitrobenzene to aniline.76b However, selectivity studies with this catalytic system were not undertaken. Reduced graphene oxide was also explored as a catalyst for hydrogenation of nitrobenzene.76c
Hydrazine hydrate-mediated reductions are much cleaner than the hydride processes as the byproducts are nitrogen and hydrogen. However, selectivity in the presence of carbon–carbon double bond, triple bond and aldehyde may be difficult to achieve, although it has been claimed in some instances.
5. In situ hydrogen generation
Decomposition of formic acid or its salts leads to evolution of CO2 gas along with H2, leaving no residual wastes. Also the CO–H2O mixture commonly known as water gas in the presence of a metal support gives CO2 and H2. This molecular H2 evolved is used for reduction (Scheme 7). Reviewed here are some recent examples exploiting this technique for nitroarene reduction (Table 5).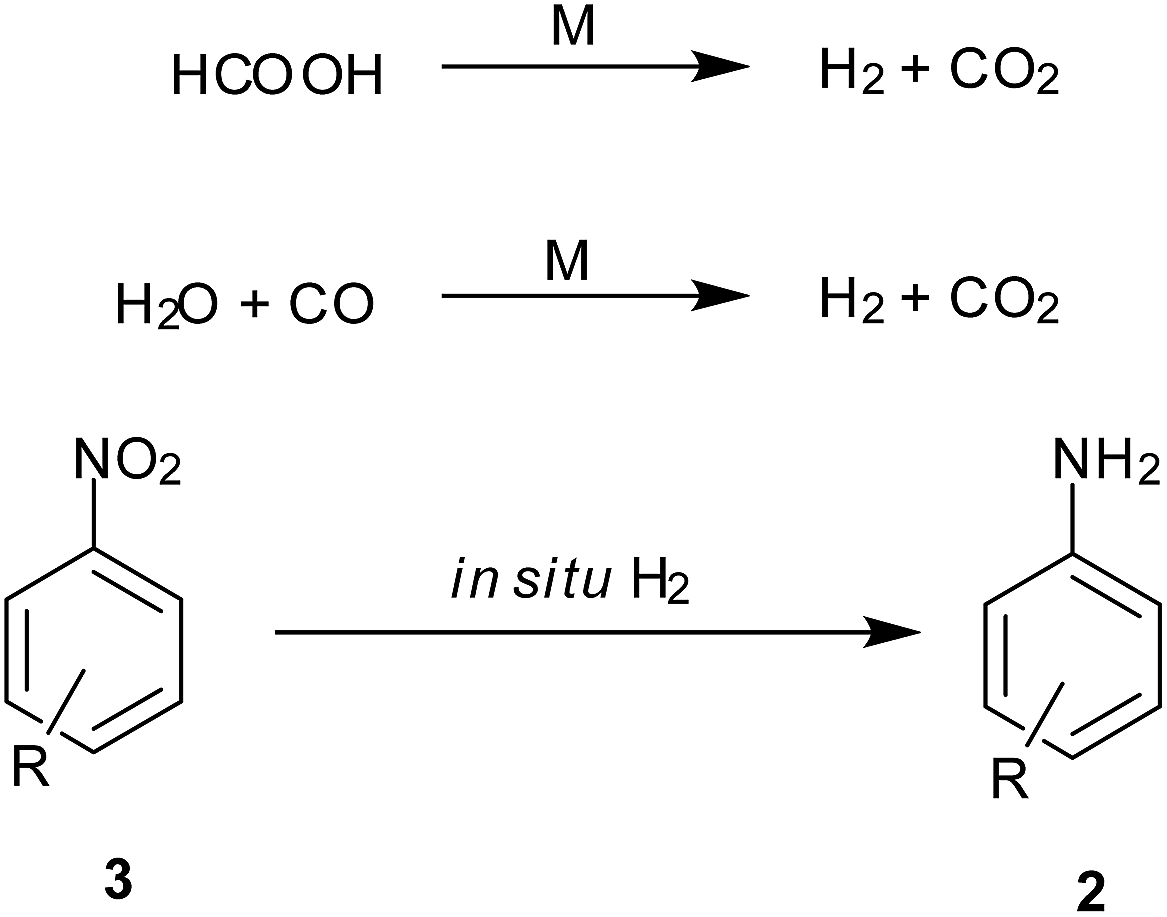 | ||
| Scheme 7 Reduction of nitroarenes by transfer hydrogenation. | ||
| Entry | Reagents (equiv.) | Solvent/conditions | Ref. |
|---|---|---|---|
| 1 | HCOOH (excess) | HTP water, 300 °C | 77 |
| 2 | CeY zeolite, HCOOH or HCOONH4 (1.6) | MW, 140 °C | 78 |
| 3 | Mo3S4H3(dmpe)3BPh4, HCOOH (3.5), Et3N | THF, 70 °C | 79 |
| 4 | Au–TiO2, CO (5 atm) | EtOH–H2O, r.t. | 80 |
| 5 | Ru–MgF2, CO (20 atm) | EtOH–H2O, 175 °C | 81 |
| 6 | 10% Pd/C, NaH2PO2 (5) | H2O, 50 °C | 82 |
| 7 | 5% Pd/C, H3PO2 (1), NaH2PO2 (3) | Ultrasound | 83 |
| 8 | H3PO3 (4)/H3PO2, NaI, aq. HBr | AcOH, 115 °C | 84 |
Continuous hydrogenation of nitrobenzene to aniline was developed by Poliakoff and coworkers in high-temperature pressurized water (HTPW) using H2 generated by thermal decomposition of HCOOH.77 This reaction is carried out in the absence of any added catalyst and can be conveniently performed on a laboratory scale.
CeY zeolite and formic acid under MW irradiation gave good yields of reduction products within 10 min. Aliphatic nitro compounds even with ester functionality were reduced to corresponding amines, while aldehyde, acid, amide, CN, Cl, Br were retained in corresponding anilines.78
Cubane-type [Mo3S4X3(dmpe)3]+ clusters have been developed as catalysts (X = H) or precatalysts (X = Cl) for the reduction of functionalized nitroarenes using formates as reducing agents.79 Functional groups like nitrile, olefin, ketone, ester, amide and even aldehyde remained intact during reduction of the nitro group.
Ru and Ir catalysts, which are not particularly selective under the conditions of conventional hydrogenation carried out with molecular hydrogen, when used in the aqueous-phase reforming/hydrogenation (APR/Hyd) process, become >99.9% selective for hydrogenation of o-CNB to o-chloroaniline.80,81
| H2PO2⊖ + 3OH⊖ → HPO32⊖ + 2H2O + 2e⊖ |
Hypophosphites are reducing agents as they get oxidized to phosphonates as shown above.82a Sodium hypophosphite was used as hydrogen source in water (containing 1% w/w Tween 20) for reduction of nitro compounds by Oba et al.82b This process was catalyzed by Pd/C (10 mol%). Aromatic as well as aliphatic nitro compounds were reduced to amines at 50 °C in more than 99% yields. Sodium hypophosphite is also used for dehalogenation, debenzylation and double bond hydrogenation. Similarly, a mixture of phosphinic acid and sodium hypophosphite with Pd/C was used as a heterogeneous catalyst in a water:2-methyl-THF system by Popowycz and coworkers.83 Here an aliphatic nitro group was selectively reduced in the presence of indole or coumarin. Nitroarenes were reduced to corresponding anilines in the presence of CN, ester, keto and halogen groups.
A novel iodide-catalyzed reduction method using hypophosphorous and/or phosphorous acids was developed by Wu et al. to reduce both diaryl ketones and nitroarenes chemoselectively in the presence of chloro and bromo substituents in high yields.84 This efficient and practical method has been successfully applied to large-scale production of a potential anticancer agent, lonafarnib.
Milder conditions and stoichiometric use of decomposing reagents and simplified workup procedures are required to make these methods popular.
6. Direct metal
Active metal can react with water to liberate hydrogen. This liberated hydrogen in the presence of metal can bring about reduction of a nitro group (Scheme 8). Also, metal could directly reduce a nitro group by electron-transfer reaction with water acting as proton source.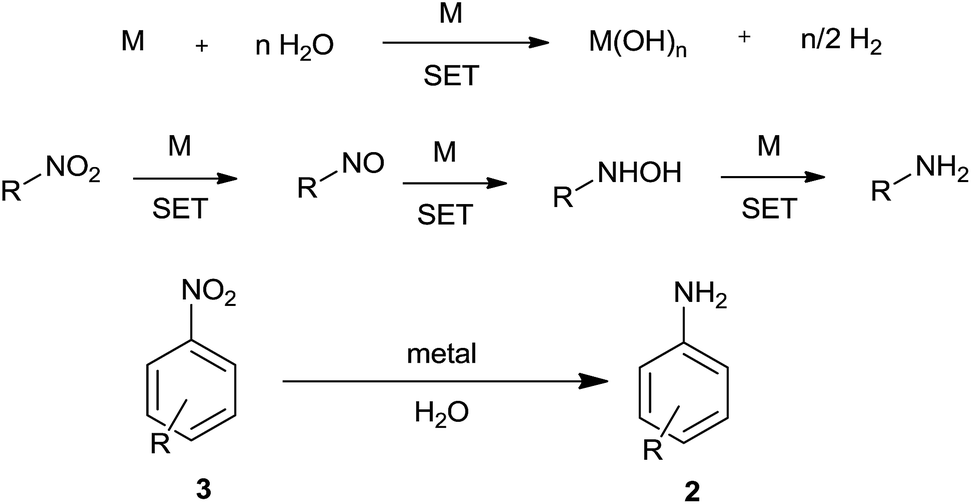 | ||
| Scheme 8 Reduction of nitroarenes using metal. | ||
 | ||
| Scheme 9 Reduction of nitroarenes by non-classical reagents. | ||
Nanosized activated metallic iron powder was used as a reducing agent by Wang et al. for reduction of nitroarenes to anilines in water at 210 °C (near critical water)85 (Table 6). This method, unlike Bechamp reduction, avoids the use of strong acidic conditions and could sustain substituents like OMe, COMe, COOEt, F, Cl, Br, and I. This method could also reduce nitronaphthalene to naphthylamine but not aliphatic nitro compounds and nitrostyrenes. Ranu and coworkers86 have achieved similarly highly selective reduction of nitroarenes using iron metal NPs in water at room temperature. During the reaction a change in shape of the Fe NPs was observed. The easily reducible functional groups CHO, COMe, CO2Me, COOH, CONH2, CN, N3, I, Br, Cl, F, SCN, O-benzyl, O-allyl, O-TBDMS, N-benzyl, and N-allyl, and styrenoid double and triple bonds were unaffected.
| Entry | Metal reagent (equiv.) | Solvent | Ref. |
|---|---|---|---|
| 1 | Fe nm powder (3) | H2O, 210 °C | 85 |
| 2 | Fe NPs (3) | H2O, r.t. | 86 |
| 3 | FeS (5), NH4Cl | MeOH, H2O, reflux | 87 |
| 4 | Te (3) | H2O, 275 °C | 88 |
| 5 | Zn (7), NH4Cl | H2O, 80 °C | 89 |
| 6 | Zn, CO2 (80 atm) | H2O, 80 °C | 90 |
| 7 | Zn, CO2 (1 atm) | H2O, r.t. | 91 |
| 8 | Zn, CO2 (1 atm) | H2O, ultrasound | 92 |
| 9 | RuCl2(PPh3)3 (0.025), KOH (0.25), Zn (3.3) | H2O, dioxane, 40 °C | 93 |
| 10 | Zn (7), SiO2–PEG | H2O, r.t. – reflux | 94 |
| 11 | Sm (2), AcOH | [BMIM][BF4], r.t. | 95 |
| 12 | Mn (2.5), CuCl2 (0.05) | THF, H2O, r.t. | 96 |
| 13 | NbCl5 (2), In (8) | THF, r.t. | 97 |
Reduction was achieved in refluxing MeOH–water mixture using FeS and ammonium chloride by Desai et al.87 Sensitive substituents like chloro, ester, and N-benzyl were unreactive in this reduction and corresponding anilines were obtained in 56 to 81% yields. Te metal was used as a reducing agent for preparation of anilines from nitroaromatics in neat critical water at 275 °C by Wang et al.88 Electron-donating (Me) and electron-withdrawing (MeCO, Cl) substituents were well tolerated. However, in the case of Br and I derivatives, competitive dehalogenation takes place. Carboxylic acid group also undergoes decarboxylation. This process does not reduce aliphatic nitro and nitrostyrenes.
Chemoselective reduction of nitroarenes to anilines was reported using Zn and NH4Cl in water at 80 °C by Tsukinoki and Tsuzuki.89a Functionalities like ester, amide and halogen were unaffected, and sterically hindered 2,6-dimethylnitrobenzene was also reduced to corresponding aniline in 95% yield. Similarly zinc powder in aqueous solutions of chelating ethers was used by Kumar and Lokanatha Rai.89b Other reducible groups like ester, chloro, amide, ketone and styryl remained unaffected. Interestingly the aliphatic nitro functionality present in 2-nitrodihydroindole also could be reduced by this method. The donor ether acts as a ligand and also serves as a co-solvent with water being the proton source. Using the commercially available designer surfactant TPGS-750-M along with Zn dust and NH4Cl, this reaction took place under mild conditions at r.t. and tolerated a wide range of functionalities. Antiarrhythmic agent procainamide was synthesized in 83% yield in two steps.89c With the Zn–H2O–CO2 system, water acted as direct hydrogen donor in supercritical CO2 as solvent.90 This method of Jiang and Dong gave excellent yields of reduction products in the presence of F, Cl, Br, I, acid and ketone functional groups.
Controlled reduction of nitroarenes to N-phenylhydroxylamine was achieved by Liu et al. using Zn in CO2/H2O system.91 An 88% yield of N-phenylhydroxylamine was obtained when 3 eq. of Zn were used in 0.1 MPa CO2 at 25 °C for 1.5 h. Using these stoichiometric conditions, dinitrobenzene was selectively reduced to m-nitro-N-phenylhydroxylamine in 99% yield. Similarly zinc in CO2–water mixture with the application of ultrasound gave excellent yields in just 60 min. Other reducible functional groups like CN, keto, Cl, and Br were not affected in these methods.92 Also alkynes, ketones, or nitro groups were chemoselectively reduced using RuCl2(Ph3P)3 as catalyst and Zn/water as stoichiometric reductant by Plietker and coworkers.93
Reduction of nitro compounds to anilines was achieved in water using zinc powder and silica gel supported PEG by Reza et al.94 The products were isolated in 68–92% yield by simple acid–base purification with retention of other substituents like NH2 and COOH, and also sensitive functionalities like CHO, Cl, and CH2Br.
Sm and AcOH in ionic liquid were used at r.t. for nitro reduction in inert atmosphere by Zheng and Zhang.95 In this system halogen, CHO, COOH, CN, and NHTs groups were unaffected and corresponding anilines were obtained in 83 to 98% yields.
Reduction of aromatic nitro compounds to anilines in THF–water mixture at r.t. using Mn as reducing agent and CuCl2 as catalyst was reported by Sarmah and Dutta.96 Nitro group was selectively reduced to NH2 in the presence of OH, NH2, Cl, COOH, ester and CN with 75–88% yield. The products were isolated in pure form by simple acid–base treatment. Similarly Yoo et al. have shown that the NbCl5/In system mediates an efficient and mild reduction of aromatic nitro compounds to the corresponding amines.97 The Br, Cl, COOCH3 and COCH3 functionalities remained unaffected.
Metal reductions as such are very selective in reducing the nitro functionality, but stoichiometric requirement of metals makes these processes unattractive.
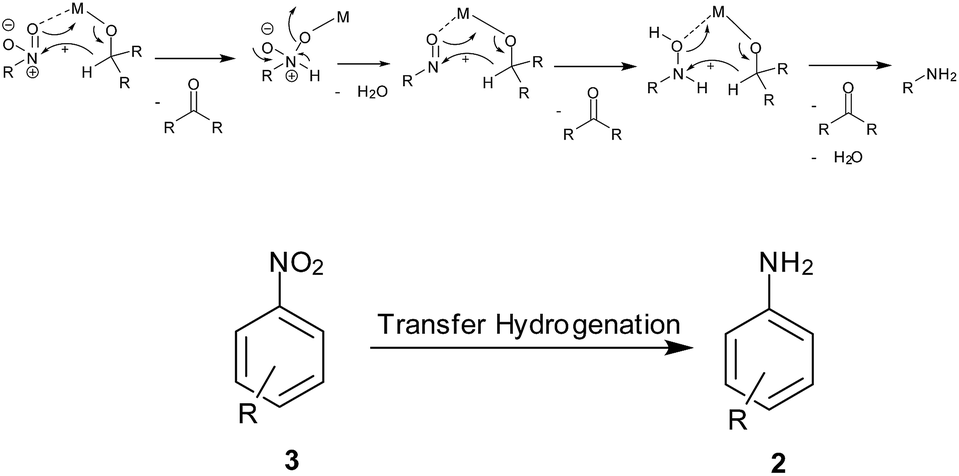
7. MPV type redox processes using organic reducing agents (transfer hydrogenation) (Scheme 9)
Perovskite-type LaFeO3 NPs were readily synthesized via thermal decomposition of the La[Fe(CN)6]·5H2O complex by Farhadi and Siadatnas98 (Table 7). This nanosized perovskite-type oxide with an average particle size of 35 nm and a specific surface area of 38.5 m2 g−1 was used as a reusable heterogeneous catalyst for selective reduction of aromatic nitro compounds into their corresponding amines by using propan-2-ol as the hydrogen donor under microwave irradiation. Chloro, bromo, nitro, ester, acid, ketone, nitrile and aldehyde groups remained intact during this process. Transfer hydrogenation of nitroaromatics to anilines in isopropyl alcohol using KOH and Ru NPs stabilized on montmorillonite clay as catalyst was achieved by Sarmah and Dutta. The catalyst was selective towards nitro reduction to corresponding anilines without affecting F, Cl, Br or CN.99a Ag-mesoporous polytriallylamine catalyst was reported under similar conditions by Salam et al.99b Recently, Fe–SBA-15 hexagonal mesopores were efficiently used for reduction of different nitro-substituted compounds using NaOH in refluxing isopropyl alcohol by Sanjini and Velmathi.99c| Entry | Reagents (equiv.) | Solvent/conditions | Ref. |
|---|---|---|---|
| 1 | LaFeO3, KOH (1), iPrOH | MW | 98 |
| 2 | Ru-acid activated montmorillonite clay or Ag–mesoporous polytriallylamine, NaOH (2.5), iPrOH | 80 °C | 99 |
| 3 | Polymer-bound palladium, K3PO4 (1.5), cyclohexanol | DMF, 110 °C | 100 |
| 4 | Ni–Fe3O4, KOH (2), glycerol | 80 °C | 101 |
| 5 | (2-Pyridyl)phenylmethanol (3.5) | Toluene, 110 °C | 102 |
| 6 | Pinacol (4), MoO2Cl2(dmf)2 | Toluene, MW, 150 °C | 103 |
| 7 | D-Glucose (2), KOH (4) | H2O:DMSO, 110 °C |
104 |
| 8 | Pd/C, 1,4-cyclohexadiene (6) | MeOH, MW, 120 °C | 105a |
| 9 | Pd13Pb9 or RhPb2 | MeOH, Ar, 70 °C | 105b |
A polymer-bound palladium catalyst was prepared in the form of PdO NPs bound on the surface of polystyrene beads by Min et al.100 This catalytic system showed good activities in the reduction of nitroarenes and the hydrodehalogenation of aryl halides with 10 mol% PdO and K3PO4 (1.5 equiv.) in DMF/cyclohexanol at 110 °C.
A heterogeneous Fe3O4–Ni magnetic NP catalyst was demonstrated for hydrogen-transfer reactions by using the environmentally friendly solvent glycerol as a hydrogen donor by Gawande et al.101
(2-Pyridyl)phenylmethanol was used as hydrogen donor for reduction of aromatic nitro compounds to arylamines. These were subsequently subjected to conjugate addition through aza-Michael reaction in a one-pot manner.102
Pinacol was used as a reducing agent in the presence of MoO2Cl2(dmf)2 as a catalyst for reduction of nitroaromatics to anilines.103 This reduction system was compatible with most halogens, amide, ester, nitriles, olefins, nitro, benzyl ether, thioether, pyridine ring and ketones. Good yields were obtained under MW conditions and acetone and water are the only by-products in this reduction. This system could also be used for deoxygenating sulfoxides.
D-Glucose, an abundantly available carbohydrate, was reported by Kumar et al. as a source of hydrogen for reduction of nitroarenes in a catalyst-free aqueous system.104 D-Glucose/KOH system in water:DMSO mixture was employed for this reduction of nitroarenes at 110 °C. Substituents like CN, CHO, CC, CN and halogens on nitroarenes were tolerated. Even dinitroarenes were found to selectively reduce to mononitroanilines in excellent yields.
Quinn et al. have shown that commonly available Pd/C or Pt/C catalyst is extremely effective with 1,4-cyclohexadiene as the hydrogen transfer source.105a For substrates containing potentially labile aromatic halogens, Pt/C is effective and results in little or no dehalogenation. In general, the reactions were complete within 5 min at 120 °C under microwave heating conditions. Furukawa and coworkers have used Pd- and Rh-based intermetallic catalysts for chemoselective catalytic transfer hydrogenation of nitro groups in styrenes, stilbenes and indoles using MeOH and 4-methylcyclohexene as hydrogen donors.105b
Transfer hydrogenations using sustainable materials under mild conditions may go a long way in meeting the future demands of such reduction processes.
8. Light-induced photocatalysis
Light induced activation of catalysts helps in reducing the energy barrier for many reactions thus providing methods with mild conditions (Scheme 10). Below are some selective reports describing the activation of passive catalysts in the presence of reducing agents to facilitate reduction under mild conditions. | ||
| Scheme 10 Reduction of nitroarenes. | ||
TiO2 was used as a photocatalyst under UV irradiation for reduction of nitrobenzene to aniline using oxalic acid as reducing agent and hole scavenger by Kominami and coworkers106 (Table 8). Vinyl, halogen, acid and ketone were unreactive in this reduction.
| Entry | Reagents | Solvent/conditions | Ref. |
|---|---|---|---|
| 1 | TiO2, Hg arc (>300 nm), oxalic acid | H2O–MeCN, r.t. | 106 |
| 2 | Ru-dye–TiO2, TEOA, 530 nm | MeCN, r.t. | 107 |
| 3 | TiO2, oxalic acid/formic acid, HCl, UV | H2O, r.t. | 108 |
| 4 | CdS nanosphere/reduced graphene oxide, 420 nm, HCOONH4 | H2O, r.t. | 109 |
| 5 | CdS, nanowires, reduced graphene oxide, >420 nm, HCOONH4 | H2O, r.t. | 110 |
| 6 | HCOONH4, Pd@CeO2, <420 nm | H2O, r.t. | 111 |
| 7 | PbBiO2Br, 440 nm, TEOA | MeCN, r.t. | 112 |
Ru dye-sensitized TiO2 was reported by Konig and coworkers as a catalyst in the presence of green light for this reduction and triethanolamine (TEOA) as reducing agent.107a Addition of a small amount of transition metals (less than 0.1 mol%) led to significant enhancement of photocatalytic activity. The optimal catalytic amount of the transition metal (Pt, Pd, Au and Ag) required for quantitative reduction depended on the nature of the metal and the method of preparation. Amounts higher than 1 mol% decreased the catalytic activity. The photocatalytic activity also depended upon the oxidation state of the metal source. Critical cluster sizes of 2 nm are required for good photocatalytic activity and the size depended upon the metal loading. Similar morphologies were found for all the transition metals. A quantum efficiency of 8% was determined for the reduction reaction under the optimized reaction conditions. Aldehyde, ketone, ester, cyano and halogen were compatible for this reduction. Dehalogenation occurs with higher loading of platinum. Green light photoreduction of nitrobenzene was also demonstrated on a laboratory preparative scale. Chen et al.107b have reported reduction of nitro compounds using TiO2 photocatalyst by UV and visible dye-sensitized systems.
Kominami and coworkers108 examined photocatalytic reduction in aqueous suspensions of titanium(IV)oxide (TiO2) in the presence of hole scavengers under various conditions. m-Nitrobenzenesulfonic acid was almost quantitatively converted into m-aminobenzenesulfonic acid in the presence of formic acid as a hole scavenger under deaerated conditions with high efficiency (>99%). Other nitroaromatic compounds were photocatalytically reduced into the corresponding amines using the same catalyst and oxalic acid. Xu and coworkers109 reported self-assembly of uniform CdS nanospheres (CdS NSPs)/graphene hybrid nanocomposites via electrostatic interaction of positively charged CdS NSPs with negatively charged graphene oxide (GO), followed by GO reduction via a hydrothermal treatment. These nanocomposites exhibited high visible light photocatalytic performance and excellent reusability toward selective reduction of aromatic nitro organics to corresponding amino organics in water in the presence of ammonium formate as a hole quencher. 2-Nitrophenol, 4-nitrophenol, 2-nitroaniline, 1-chloro-4-nitrobenzene, 4-nitroanisole and 1-bromo-4-nitrobenzene were successfully reduced to their amines without affecting the other groups present on the benzene ring. As during reduction graphene and CdS are not affected, the catalyst system can be potentially recycled. Similarly, CdS nanowires–reduced graphene oxide nanocomposites (CdS NWs–RGO NCs) were synthesized by the same process in the same laboratory. Furthermore, the presence of RGO also improves the adsorption capacity of CdS NWs–RGO NCs toward aromatic nitro organics.110
Pd NP cores encapsulated within CeO2 hollow shells were used for thermocatalytic and photocatalytic reduction of aromatic nitro compounds to anilines in water at room temperature by Zhang and Xu.111 The thermocatalytic method uses NaBH4 as reducing agent whereas the photocatalytic method uses ammonium oxalate as reducing agent and visible light irradiation. This catalyst showed good selectivity for nitro reduction in the presence of Cl and Br.
PbBiO2Cl and PbBiO2Br were used as catalysts for reduction of nitrobenzene derivatives using TEOA with blue light by Konig's group.112 The catalysts were selective for nitro reduction in the presence of CN, CHO, and keto groups but could reduce pyridinealdehyde. The catalyst could be reused many times after sonication to remove the passivity.
Direct sunlight-mediated photochemical reductions on a large scale particularly for environmental cleaning will be of great help in the future.
9. Biotic reduction
Although reduction of nitroarenes has been considered as a synthetic process so far, here are some of the reports that consider the transformation of nitroarene to anilines as a part of biological processes (Scheme 11). | ||
| Scheme 11 Reduction of nitroarenes. | ||
Mercier et al. observed that Escherichia coli is able to reduce azo compounds such as methyl red (MR) and nitro compounds such as 7-nitrocoumarin-3-carboxylic acid (7NCCA) (Table 9). An in-depth study revealed that enzyme AzoR could reduce both MR and 7NCCA, whereas enzymes NfsA and NfsB could only reduce the nitro compound.113 Similarly, a series of aliphatic and aromatic aldehydes and ketones, as well as some nitro compounds were reduced using whole plant cells from Lens culinaris seeds by Ferreira et al.114
| Entry | Natural sources | Conditions | Ref. |
|---|---|---|---|
| 1 | Escherichia coli reductases | pH 7 buffer, 30 °C | 113 |
| 2 | Plant cells from Lens culinaris seeds | H2O, 30 °C | 114 |
| 3 | Plant cells from grapes (Vitis vinifera L.) | H2O, 25 °C | 115 |
| 4 | Cattle tick Boophilus microplus, spider Nephila plumipes | In vivo | 116 |
| 5 | Microbial consortium | H2, pH 6.5–6.8, 30 °C | 117 |
| 6 | Biocatalyzed cathode | Glucose, 25 °C | 118 |
| 7 | FMN-dependent nitro-reductase | Glucose | 119 |
| 8 | BaNTR1, BmGDH NADP | Glucose, 0.1 M sodium phosphate buffer, 30 °C | 120 |
Plant cells from a grape (Vitis vinifera L.) reducing aromatic nitro compounds under mild conditions to the corresponding hydroxylamines was observed by Li et al.115
Two species of Arachnida, Boophilus microplus (cattle tick) and Nephila plumipes (Sydney spider), metabolized 14C nitrobenzene to aniline in vivo. These species could also metabolize N,N-dimethylaminoazobenzene to anilines. This was the first and only report of observing reduction of nitrobenzene to aniline in living organisms by Holder and Willox.116
Conversion of nitrobenzene to aniline, a less toxic end product that can easily be mineralized, was carried out in a continuous-flow anaerobic bioreactor using H2 gas and a microbial consortium by Cao et al.117 This reduction is sensitive to both pH and temperature. Optimum reduction was obtained at pH 6.5–6.8 and at 30 °C.
A fed-batch bioelectrochemical system with a microbial catalyzed cathode could transform nitrobenzene to aniline within 24 h when a voltage of 0.5 V was applied in the presence of glucose, as reported by Wang et al.118
FMN-dependent ene-reductases and nitroreductases can catalyze or mediate a diverse spectrum of chemical reactions due to the chemical versatility of the flavin cofactor. Nitroreductases have evolved as natural remediation tools in contaminated environments with a major role in the reduction of toxic nitroaromatics.119
Bacterial nitroreductase BaNTR1 has recently been identified and used as biocatalyst by Xu and coworkers for controllable reduction of nitroarenes with electron-withdrawing groups like NO2, CN, amide, acid and ester to corresponding N-arylhydroxylamines.120
Enzymatic reductions have shown great promise, and sustained research in this field is required for future developments.
10. Conclusions
The area of research involving methods for the reduction of nitroarenes continues to attract synthetic chemists due to problems associated with selectivity, cost of process, ease of reaction and the benignity involved. The market potential for a new industrial application is also very high due to the demand of the final reduction product, aniline. With the rapidly developing nanotechnology, ever newer materials are being generated and these newly generated nanomaterials may lead to more selective and more efficient processes. Of the newly tried metals, gold has shown great promise and it may be a choice of metal replacing traditional palladium and platinum metals. Further progress in this field is expected particularly using magnetic nanocomposites, which can be recycled easily. Research into non-coinage metals will continue to take place due to the cost factor involved in the noble metal reduction processes. Cost-effective green alternatives of transfer hydrogenation, enzymatic and photochemical reduction methods are the ones where more progress is expected.Acknowledgements
The authors acknowledge the Council of Scientific and Industrial Research (CSIR), University Grants Commission (UGC) and Department of Science and Technology (DST, nano mission), New Delhi for financial assistance.References
- (a) J. Andraos and A. P. Dicks, Chem. Educ. Res. Pract., 2012, 13, 69 RSC; (b) A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1 RSC; (c) R. Mestres, Green Chem., 2004, G10 RSC; (d) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC; (e) R. A. Sheldon, Green Chem., 2008, 10, 359 RSC; (f) P. J. Dunn, Chem. Soc. Rev., 2012, 41, 1452 RSC; (g) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC; (h) P. Anatas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (i) E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038 RSC; (j) S. Y. Oh and P. C. Chiu, Environ. Sci. Technol., 2009, 43, 6983 CrossRef CAS.
- (a) P. J. Dunn, Green Chem., 2013, 15, 3099 RSC; (b) R. L. Lankey and P. T. Anastas, Green Chem., 2000, 2, 289 RSC; (c) J. H. Clark, Green Chem., 2006, 8, 17 RSC; (d) R. A. Sheldon, Chem. Commun., 2008, 3352 RSC; (e) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (f) W. J. W. Watson, Green Chem., 2012, 14, 251 RSC.
- (a) V. Pandarus, R. Ciriminna, F. Béland and M. Pagliaro, Adv. Synth. Catal., 2011, 353, 1306 CrossRef CAS PubMed; (b) Y. Mikami, A. Noujima, T. Mitsudome, T. Mizugaki, K. Jitsukawa and K. Kaneda, Chem. Lett., 2010, 39, 223 CrossRef CAS; (c) F. Cárdenas-Lizana, S. Gómez-Quero and M. A. Keane, Catal. Commun., 2008, 9, 475 CrossRef PubMed.
- (a) T. Tsukinoki and H. Tsuzuki, Green Chem., 2001, 3, 37 RSC; (b) B. Sreedhar, D. K. Devi and D. Yada, Catal. Commun., 2011, 12, 1009 CrossRef CAS PubMed; (c) V. Mohan, C. V. Pramod, M. Suresh, K. H. P. Reddy, B. David Raju and K. S. Rama Rao, Catal. Commun., 2012, 18, 89 CrossRef CAS PubMed.
- (a) F. Ellis, Paracetamol: a curriculum resource, Royal Society of Chemistry, Cambridge, 2002 Search PubMed; (b) A. S. Travis, Manufacture and uses of the anilines: a vast array of processes and products, The chemistry of Anilines Part 1, Wiley, 2009, p. 764 Search PubMed.
- (a) P. F. Schellhammer, Expert Opin. Pharmacother., 2002, 3, 1313 CrossRef CAS PubMed; (b) Y. Fradet, N. James and J. Maher, Expert Rev. Anticancer Ther., 2004, 4, 37 CrossRef CAS; (c) W. A. See and C. J. Tyrrell, J. Cancer Res. Clin. Oncol., 2006, 132 Search PubMed; (d) I. I. Müderris, F. Bayram, B. Ozçelik and M. Güven, Gynecol. Endocrinol., 2002, 16, 63 CrossRef PubMed.
- (a) W. Kassouf, S. Tanguay and A. G. Aprikian, J. Urol., 2003, 169, 1742 CrossRef CAS PubMed; (b) M. Moguilewsky, C. Bertagna and M. Hucher, J. Steroid Biochem., 1987, 871 CrossRef CAS; (c) A. C. Hsieh and C. J. Ryan, Cancer J., 2008, 14, 11 CrossRef CAS PubMed.
- (a) Z. Li, M. Xu, S. Xing, W. Ho, T. Ishii, Q. Li, X. Fu and Z. Zhao, J. Biol. Chem., 2007, 282, 3428 CrossRef CAS PubMed; (b) A. Dudek, K. Kmak, J. Koopmeiners and M. Keshtgarpour, Lung Cancer, 2006, 51, 89 CrossRef PubMed.
- (a) S. J. Brickner, Curr. Pharm. Des., 1996, 2, 175 CAS; (b) G. Y. Xu, Y. Zhou and M. C. Xu, Chin. Chem. Lett., 2006, 17, 302 CAS; (c) B. B. Lohray, S. Baskaran, B. S. Rao, B. Y. Reddy and I. N. Rao, Tetrahedron Lett., 1999, 40, 4855 CrossRef CAS.
- J. Eron, P. Yeni, J. Gathe, V. Estrada, E. DeJesus, S. Staszewski, P. Lackey, C. Katlama, B. Young, L. Yau, D. S. Phillips, P. Wannamaker, C. Vavro, L. Patel, J. Yeo and M. Shaefer, Lancet, 2006, 368, 476 CrossRef CAS.
- (a) CAS Scifinder® summarizes around 500 research reports for past 5 years (2009–2014) on the topic “Reduction of Nitrobenzene”; (b) A. M. Tafesh and J. Weiguny, Chem. Rev., 1996, 96, 2035 CrossRef CAS PubMed; (c) H. U. Blaser, H. Steiner and M. Studer, ChemCatChem, 2009, 1, 210 CrossRef CAS PubMed; (d) P. Lara and K. Philippot, Catal. Sci. Technol., 2014, 4, 2445 RSC.
- (a) J. Pan, J. Liu, S. Guo and Z. Yang, Catal. Lett., 2009, 131, 179 CrossRef CAS; (b) X. B. Lou, L. He, Y. Qian, Y. M. Liu, Y. Cao and K. N. Fan, Adv. Synth. Catal., 2011, 353, 281 CrossRef CAS PubMed; (c) K. V. R. Chary and C. S. Srikanth, Catal. Lett., 2009, 128, 164 CrossRef CAS; (d) R. J. Kalbasi, A. A. Nourbakhsh and F. Babaknezhad, Catal. Commun., 2011, 12, 955 CrossRef CAS PubMed; (e) U. Sharma, P. Kumar, N. Kumar, V. Kumar and B. Singh, Adv. Synth. Catal., 2010, 352, 1834 CrossRef CAS PubMed; (f) R. V. Jagadeesh, G. Wienhofer, F. A. Westerhaus, A. E. Surkus, M. M. Pohl, H. Junge, K. Junge and M. Beller, Chem. Commun., 2011, 47, 10972 RSC; (g) G. Wienhofer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875 CrossRef PubMed; (h) Z. Zhao, H. Yang, Y. Li and X. Guo, Green Chem., 2014, 16, 1274 RSC; (i) F. A. Westerhaus, R. V. Jagadeesh, G. Wienhöfer, M. M. Pohl, J. Radnik, A. E. Surkus, J. Rabeah, K. Junge, H. Junge, M. Nielsen, A. Brückner and M. Beller, Nat. Chem., 2013, 5, 537 CrossRef CAS PubMed; (j) H. I. Schlesinger, H. C. Brown, E. Finholt, J. R. Gilbreath, H. R. Hoekstra and E. Hyde, J. Am. Chem. Soc., 1953, 75, 215 CrossRef CAS; (k) U. Leutenegger, A. Madin and A. Pfaltz, Angew. Chem., Int. Ed., 1989, 28, 60 CrossRef PubMed; (l) M. F. Lv, G. P. Lu and C. Cai, Asian J. Org. Chem., 2015, 4, 141 CrossRef CAS PubMed; (m) Q. Ge, J. Ran, L. Wu and T. Xu, J. Appl. Polym. Sci., 2014, 132, 41268 Search PubMed.
- (a) F. C. Lizana, D. Lamey, S. G. Quero, N. Perret, L. K. Minsker and M. A. Keane, Catal. Today, 2011, 173, 53 CrossRef PubMed; (b) X. Wang, M. Liang, J. Zhang and Y. Wang, Curr. Org. Chem., 2007, 11, 299 CrossRef CAS.
- A. Béchamp, Ann. Chim. Phys., 1854, 42, 186 Search PubMed.
- (a) H. K. Kadam, S. Khan, R. Kunkalkar and S. G. Tilve, Tetrahedron Lett., 2013, 54, 1003 CrossRef CAS PubMed; (b) H. K. Kadam and S. G. Tilve, RSC Adv., 2012, 2, 6057 RSC; (c) A. A. Vernekar, S. Patil, C. Bhat and S. G. Tilve, RSC Adv., 2013, 3, 13243 RSC.
- F. Zhang, J. Jin, X. Zhong, S. Li, J. Niu, R. Li and J. Ma, Green Chem., 2011, 13, 1238 RSC.
- (a) A. Amali and R. K. Rana, Green Chem., 2009, 11, 1781 RSC; (b) R. Zhang, J. Liu, F. Li, S. Wang, C. Xia and W. Sun, Chin. J. Chem., 2011, 29, 525 CrossRef CAS PubMed.
- H. Ji, Q. Long, Y. He and X. Yao, Sci. China: Chem., 2010, 53, 1520 CrossRef CAS.
- M. Chatterjee, T. Ishizaka, T. Suzuki, A. Suzuki and H. Kawanami, Green Chem., 2012, 14, 3415 RSC.
- B. Sreedhar, D. Devi and D. Yada, Catal. Commun., 2011, 12, 1009 CrossRef CAS PubMed.
- (a) X. Han and J. Li, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2007, 46, 1747 Search PubMed; (b) Z. Sun, Y. Zhao, Y. Xie, R. Tao, H. Zhang, C. Huang and Z. Liu, Green Chem., 2010, 12, 1007 RSC.
- P. Lara, A. Suarez, V. Colliere, K. Philippot and B. Chaudret, ChemCatChem, 2014, 6, 87 CrossRef CAS PubMed.
- X. Yuan, N. Yan, C. Xiao, C. Li, Z. Fei, Z. Cai, Y. Kou and P. J. Dyson, Green Chem., 2010, 12, 228 RSC.
- Y. Motoyama, K. Kamo and H. Nagashima, Org. Lett., 2009, 11, 1345 CrossRef CAS PubMed.
- Y. Takenaka, T. Kiyosu, J. C. Choi, T. Sakakura and H. Yasuda, Green Chem., 2009, 11, 1385 RSC.
- (a) A. Corma and P. Serna, Nat. Protoc., 2007, 1, 2590 CrossRef PubMed; (b) A. Grirrane, A. Corma and H. Garcia, Nat. Protoc., 2010, 5, 429 CrossRef CAS PubMed.
- X. Tan, Z. Zhang, Z. Xiao, Q. Xu, C. Liang and X. Wang, Catal. Lett., 2012, 142, 788 CrossRef CAS.
- D. He, H. Shi, Y. Wu and B. Xu, Green Chem., 2007, 9, 849 RSC.
- Y. Matsushima, R. Nishiyabu, N. Takanashi, M. Haruta, H. Kimura and Y. Kubo, J. Mater. Chem., 2012, 22, 24124 RSC.
- D. He, X. Jiao, P. Jiang, J. Wang and B. Xu, Green Chem., 2012, 14, 111 RSC.
- T. Mitsudome, Y. Mikami, M. Matoba, T. Mizugaki, K. Jitsukawa and K. Kaneda, Angew. Chem., 2012, 51, 136 CrossRef CAS PubMed.
- M. A. Harrad, B. Boualy, L. Firdoussi, A. Mehdi, C. Santi, S. Giovagnoli, M. Nocchetti and M. Ali, Catal. Commun., 2013, 32, 92 CrossRef CAS PubMed.
- X. Meng, H. Cheng, S. Fujita, Y. Yu, F. Zhao and M. Arai, Green Chem., 2011, 13, 570 RSC.
- (a) Y. Zheng, K. Ma, H. Wang, X. Sun, J. Jiang, C. Wang, R. Li and J. Ma, Catal. Lett., 2008, 124, 268 CrossRef CAS; (b) W. J. Liu, K. Tian and H. Jiang, Green Chem., 2015, 17, 821 RSC.
- G. Fan, W. Huang and C. Wang, Nanoscale, 2013, 5, 6819 RSC.
- R. F. D'Vries, M. Iglesias, N. Snejko, S. Alvarez-Garcia, E. Gutierrez-Pueblaa and M. A. Monge, J. Mater. Chem., 2012, 22, 1191 RSC.
- (a) S. Harish, J. Mathiyarasu, K. L. N. Phani and V. Yegnaraman, Catal. Lett., 2009, 128, 197 CrossRef CAS; (b) A. K. Shil, D. Sharma, N. R. Guha and P. Das, Tetrahedron Lett., 2012, 53, 4858 CrossRef CAS PubMed.
- P. K. Verma, M. Bala, K. Thakur, U. Sharma, N. Kumar and B. Singh, Catal. Lett., 2014, 144, 1258 CrossRef CAS.
- (a) F. Lin and R. Doong, J. Phys. Chem. C, 2011, 115, 6591 CrossRef CAS; (b) R. K. Sharma, Y. Monga and A. Puri, J. Mol. Catal. A: Chem., 2014, 393, 84 CrossRef CAS PubMed.
- K. Layek, M. LakshmiKantam, M. Shirai, D. N. Hamane, T. Sasaki and H. Maheswaran, Green Chem., 2012, 14, 3164 RSC.
- X. Bai, Y. Gao, H. Liu and L. Zheng, J. Phys. Chem. C, 2009, 113, 17730 CAS.
- H. Wu, X. Huang, M. Gao, X. Liao and B. Shi, Green Chem., 2011, 13, 651 RSC.
- Y. Yao, Y. Sun, Y. Han and C. Yan, Chin. J. Chem., 2010, 28, 705 CrossRef CAS PubMed.
- D. M. Dotzauer, S. Bhattacharjee, Y. Wen and M. L. Bruening, Langmuir, 2009, 25, 1865 CrossRef CAS PubMed.
- E. Seo, J. Kim, Y. Hong, Y. S. Kim, D. Lee and B. Kim, J. Phys. Chem. C, 2013, 117, 11686 CAS.
- J. Li, C. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426 RSC.
- E. Vasilikogiannaki, C. Gryparis, V. Kotzabasaki, I. N. Lykakis and M. Stratakis, Adv. Synth. Catal., 2013, 355, 907 CrossRef CAS PubMed.
- P. Liu and M. Zhao, Appl. Surf. Sci., 2009, 255, 3989 CrossRef CAS PubMed.
- A. Leelavathi, T. U. B. Rao and T. Pradeep, Nanoscale Res. Lett., 2011, 6, 123 CrossRef PubMed.
- Q. An, M. Yu, Y. Zhang, W. Ma, J. Guo and C. Wang, J. Phys. Chem. C, 2012, 116, 22432 CAS.
- R. Rajesh and R. Venkatesan, J. Mol. Catal. A: Chem., 2012, 359, 88 CrossRef CAS PubMed.
- R. Vadakkekara, M. Chakraborty and P. A. Parikh, Colloids Surf., A, 2012, 399, 11 CrossRef CAS PubMed.
- S. Wunder, F. Polzer, Y. Lu, Y. Mei and M. Ballauff, J. Phys. Chem. C, 2010, 114, 8814 CAS.
- (a) R. Kaur, C. Giordano, M. Gradzielski and S. K. Mehta, Chem.–Asian J., 2014, 9, 189 CrossRef CAS PubMed; (b) H. Zhang, S. Gao, N. Shang, C. Wang and Z. Wang, RSC Adv., 2014, 4, 31328 RSC; (c) Y. S. Feng, J. J. Ma, Y. M. Kang and H. J. Xu, Tetrahedron, 2014, 70, 6100 CrossRef CAS PubMed; (d) R. Kaur and B. Pal, Appl. Catal., A, 2015, 491, 28 CrossRef CAS PubMed; (e) Y. Feng, A. Wang, H. Yin, X. Yan and L. Shen, Chem. Eng. J., 2015, 262, 427 CrossRef CAS PubMed.
- F. Wu, L. Qiu, F. Ke and X. Jiang, Inorg. Chem. Commun., 2013, 32, 5 CrossRef CAS PubMed.
- S. Pina Jr, D. M. Cedillo, C. Tamez, N. Izquierdo, J. G. Parsons and J. J. Gutierrez, Tetrahedron Lett., 2014, 55, 5468 CrossRef PubMed.
- (a) J. Lipowitz and S. A. Bowman, J. Org. Chem., 1973, 38, 162 CrossRef CAS; (b) R. J. Rahaim and R. E. Maleczka, Org. Lett., 2005, 7, 5087 CrossRef CAS PubMed.
- (a) R. G. de Noronha, C. C. Romao and A. C. Fernandes, J. Org. Chem., 2009, 74, 6960 CrossRef CAS PubMed; (b) H. R. Brinkman, W. H. Miles, M. D. Hilborn and M. C. Smith, Synth. Commun., 1996, 26, 973 CrossRef CAS PubMed.
- (a) L. Pehlivan, E. Métay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron Lett., 2010, 51, 1939 CrossRef CAS PubMed; (b) L. Pehlivan, E. Metay, S. Laval, W. Dayoub, P. Demonchaux, G. Mignani and M. Lemaire, Tetrahedron, 2011, 67, 1971 CrossRef CAS PubMed.
- K. Junge, B. Wendt, N. Shaikh and M. Beller, Chem. Commun., 2010, 46, 1769 RSC.
- S. Park, I. S. Lee and J. Park, Org. Biomol. Chem., 2013, 11, 395 CAS.
- V. Yadav, S. Gupta, R. Kumar, G. Singh and R. Lagarkha, Synth. Commun., 2012, 42, 213 CrossRef CAS PubMed.
- (a) Y. M. Lu, H. Z. Zhu, W. G. Li, B. Hu and S. H. Yu, J. Mater. Chem. A, 2013, 1, 3783 RSC; (b) F. Lia, B. Fretta and H. Li, Synlett, 2014, 25, 1403 CrossRef.
- (a) S. Kim, E. Kim and B. Moon Kim, Chem.–Asian J., 2011, 6, 1921 CrossRef CAS PubMed; (b) D. Cantillo, M. M. Moghaddam and C. O. Kappe, J. Org. Chem., 2013, 78, 4530 CrossRef CAS PubMed.
- (a) H. Zhang, C. Feng, N. Shang, S. Gao, C. Wang and Z. Wang, Lett. Org. Chem., 2013, 10, 17 CAS; (b) C. Feng, H. Zhang, N. Shang, S. Gao and C. Wang, Chin. Chem. Lett., 2013, 24, 539 CrossRef CAS PubMed; (c) M. Shokouhimehr, T. Kim, S. W. Jun, K. Shin, Y. Jang, B. H. Kim, J. Kim and T. Hyeon, Appl. Catal., A, 2014, 476, 133 CrossRef CAS PubMed.
- Q. Shi, R. Lu, K. Ji, Z. Zhang and D. Zhao, Green Chem., 2006, 8, 868 RSC.
- U. Sharma, P. K. Verma, N. Kumar, V. Kumar, M. Bala and B. Singh, Chem.–Eur. J., 2011, 17, 5903 CrossRef CAS PubMed.
- Y. Jang, S. Kim, S. W. Jun, B. H. Kim, S. Hwang, I. K. Song, B. M. Kim and T. Hyeon, Chem. Commun., 2011, 47, 3601 RSC.
- M. Shokouhimehr, J. E. Lee, S. Ihn Han and T. Hyeon, Chem. Commun., 2013, 49, 4779 RSC.
- P. Luo, K. Xu, R. Zhang, L. Huang, J. Wang, W. Xing and J. Huang, Catal. Sci. Technol., 2012, 2, 301 CAS.
- U. Sharma, N. Kumar, P. K. Verma, V. Kumar and B. Singh, Green Chem., 2012, 14, 2289 RSC.
- B. Raju, R. Ragul and B. N. Sivasankar, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2009, 48, 1315 Search PubMed.
- A. V. Orosz and L. Marko, Transition Met. Chem., 1988, 13, 221 CrossRef.
- L. Huang, P. Luo, M. Xiong, R. Chen, Y. Wang, W. Xing and J. Huang, Chin. J. Chem., 2013, 31, 987 CrossRef CAS PubMed.
- (a) R. K. Rai, A. Mahata, S. Mukhopadhyay, S. Gupta, P. Z. Li, K. T. Nguyen, Y. Zhao, B. Pathak and S. K. Singh, Inorg. Chem., 2014, 53, 2904 CrossRef CAS PubMed; (b) Z. Zhao, H. Yang, Y. Li and X. Guo, Green Chem., 2014, 16, 1274 RSC; (c) J. W. Larsen, M. Freund, K. Y. Kim, M. Sidovar and J. L. Stuart, Carbon, 2000, 38, 655 CrossRef CAS.
- (a) X. Gu, W. Qi, S. Wu, Z. Sun, X. Xu and D. Su, Catal. Sci. Technol., 2014, 4, 1730 RSC; (b) P. Tang, G. Hu, Y. Gao, W. Li, S. Yao, Z. Liu and D. Ma, Sci. Rep., 2014, 4, 5901 CAS; (c) Y. Gao, D. Ma, C. Wang, J. Guan and X. Bao, Chem. Commun., 2011, 47, 2432 RSC.
- E. G. Verdugo, Z. Liu, E. Ramirez, J. G. Serna, J. F. Dubreuil, J. R. Hyde, P. A. Hamley and M. Poliakoff, Green Chem., 2006, 8, 359 RSC.
- K. Arya and A. Dandia, J. Korean Chem. Soc., 2010, 54, 55 CrossRef CAS.
- I. Sorribes, G. Wienhofer, C. Vicent, K. Junge, R. Llusar and M. Beller, Angew. Chem., Int. Ed., 2012, 51, 7794 CrossRef CAS PubMed.
- (a) L. He, L. Wang, H. Sun, J. Ni, Y. Cao, H. He and K. Fan, Angew. Chem., 2009, 121, 9702 CrossRef PubMed; (b) L. He, L. Wang, H. Sun, J. Ni, Y. Cao, H. He and K. Fan, Angew. Chem., Int. Ed., 2009, 48, 9538 CrossRef CAS PubMed.
- M. Pietrowski, Green Chem., 2011, 13, 1633 RSC.
- (a) N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, 2nd edn, 1997 Search PubMed; (b) M. Oba, K. Kojima, M. Endo, H. Sano and K. Nishima, Green Chem. Lett. Rev., 2013, 6, 233 CrossRef CAS PubMed.
- M. Baron, E. Metay, M. Lemaire and F. Popowycz, Green Chem., 2013, 15, 1006 RSC.
- G. G. Wu, F. X. Chen, D. LaFrance, Z. Liu, S. G. Greene, Y. Wong and J. Xie, Org. Lett., 2011, 13, 5220 CrossRef CAS PubMed.
- L. Wang, P. Li, Z. Wu, J. Yan, M. Wang and Y. Ding, Synthesis, 2003, 13, 2001 CrossRef.
- R. Dey, N. Mukherjee, S. Ahammed and B. C. Ranu, Chem. Commun., 2012, 48, 7982 RSC.
- D. G. Desai, S. S. Swami, S. K. Dabhade and M. G. Ghagare, Synth. Commun., 2001, 31, 1249 CrossRef CAS PubMed.
- L. Wang, P. H. Li and Z. Q. Jiang, Chin. J. Chem., 2003, 21, 222 CAS.
- (a) T. Tsukinoki and H. Tsuzuki, Green Chem., 2001, 3, 37 RSC; (b) P. S. Kumar and K. M. Lokanatha Rai, Chem. Pap., 2012, 66, 772 CAS; (c) S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98 CrossRef CAS PubMed.
- H. F. Jiang and Y. S. Dong, Chin. J. Chem., 2008, 26, 1407 CrossRef CAS PubMed.
- S. Liu, Y. Wang, J. Jiang and Z. Jin, Green Chem., 2009, 11, 1397 RSC.
- S. Liu, Y. Wang, X. Yang and J. Jiang, Res. Chem. Intermed., 2012, 38, 2471 CrossRef CAS.
- T. Schabel, C. Belger and B. Plietker, Org. Lett., 2013, 15, 2858 CrossRef CAS PubMed.
- K. A. Reza, Z. Maryam, M. T. Fatemeh and F. M. Mehdi, Iran. J. Chem. Chem. Eng., 2011, 30, 37 Search PubMed.
- X. L. Zheng and Y. M. Zhang, Chin. J. Chem., 2002, 20, 925 CrossRef CAS PubMed.
- P. Sarmah and D. K. Dutta, J. Chem. Res., 2003, 236 CrossRef CAS.
- B. W. Yoo, D. Kim, H. M. Kim and S. H. Kang, Bull. Korean Chem. Soc., 2012, 33, 2851 CrossRef CAS.
- S. Farhadi and F. Siadatnas, J. Mol. Catal. A: Chem., 2011, 339, 108 CrossRef CAS PubMed.
- (a) P. P. Sarmah and D. K. Dutta, Green Chem., 2012, 14, 1086 RSC; (b) N. Salam, B. Banerjee, A. S. Roy, P. Mondal, S. Roy, A. Bhaumik and M. Islam, Appl. Catal., A, 2014, 477, 184 CrossRef CAS PubMed; (c) N. S. Sanjini and S. Velmathi, RSC Adv., 2014, 4, 15381 RSC.
- H. Min, S. Lee, M. Park, J. Hwang, H. M. Jung and S. Lee, J. Organomet. Chem., 2014, 755, 7 CrossRef CAS PubMed.
- M. B. Gawande, A. K. Rathi, P. S. Branco, I. D. Nogueira, A. Velhinho, J. J. Shrikhande, U. U. Indulkar, R. V. Jayaram, C. Ghumman, N. Bundaleski and O. Teodoro, Chem.–Eur. J., 2012, 18, 12628 CrossRef CAS PubMed.
- D. Giomi, R. Alfini and A. Brandi, Tetrahedron, 2011, 67, 167 CrossRef CAS PubMed.
- N. Garcia, P. G. Garcia, M. A. Rodriguez, R. Rubio, M. R. Pedrosa, F. J. Arnaiz and R. Sanz, Adv. Synth. Catal., 2012, 354, 321 CrossRef CAS PubMed.
- M. Kumar, U. Sharma, S. Sharma, V. Kumar, B. Singh and N. Kumar, RSC Adv., 2013, 3, 4894 RSC.
- (a) J. F. Quinn, C. E. Bryant, K. C. Golden and B. T. Gregg, Tetrahedron Lett., 2010, 51, 786 CrossRef CAS PubMed; (b) S. Furukawa, Y. Yoshida and T. Komatsu, ACS Catal., 2014, 4, 1441 CrossRef CAS.
- K. Imamura, K. Hashimoto and H. Kominami, Chem. Commun., 2012, 48, 4356 RSC.
- (a) S. Fuldner, R. Mild, H. Siegmund, J. Schroeder, M. Gruber and B. Konig, Green Chem., 2010, 12, 400 RSC; (b) S. Chen, H. Zhang, X. Yu and W. Liu, Chin. J. Chem., 2011, 29, 399–404 CrossRef CAS PubMed.
- K. Imamura, S. Iwasaki, T. Maeda, K. Hashimoto, B. Ohtani and H. Kominami, Phys. Chem. Chem. Phys., 2011, 13, 5114 RSC.
- Z. Chen, S. Liu, M. Yang and Y. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 4309 CAS.
- (a) S. Liu, Z. Chen, N. Zhang, Z. Tang and Y. Xu, J. Phys. Chem. C, 2013, 117, 8251 CrossRef CAS; (b) X. Dai, M. Xie, S. Meng, X. Fu and S. Chen, Appl. Catal., B, 2014, 158–159, 382 CrossRef CAS PubMed.
- N. Zhang and Y. Xu, Chem. Mater., 2013, 25, 1979 CrossRef CAS.
- S. Fuldner, P. Pohla, H. Bartling, S. Dankesreiter, R. Stadler, M. Gruber, A. Pfitzner and B. Konig, Green Chem., 2011, 13, 640 RSC.
- C. Mercier, V. Chalansonnet, S. Orenga and C. Gilbert, J. Appl. Microbiol., 2013, 115, 1012 CAS.
- D. A. Ferreira, R. Silva, J. Assunçao, M. Mattos, T. Lemos and F. Monte, Biotechnol. Bioprocess Eng., 2012, 17, 407 CrossRef CAS.
- F. Li, J. Cui, X. Qian, R. Zhanga and Y. Xiao, Chem. Commun., 2005, 1901 RSC.
- G. M. Holder and S. Willox, Life Science, 1973, 13, 391 CrossRef CAS.
- H. B. Cao, Y. P. Li, G. F. Zhang and Y. Zhang, Biotechnol. Lett., 2004, 26, 307 CrossRef CAS.
- A. Wang, H. Cheng, B. Liang, N. Ren, D. Cui, N. Lin, B. H. Kim and K. Rabaey, Environ. Sci. Technol., 2011, 45, 10186 CrossRef CAS PubMed.
- K. Durchschein, M. Hall and K. Faber, Green Chem., 2013, 15, 1764 RSC.
- H. Nguyen, G. Zheng, X. Qian and J. Xu, Chem. Commun., 2014, 50, 2861 RSC.
| This journal is © The Royal Society of Chemistry 2015 |