
Influence of Ti4+ on the long lasting luminescence of Sr2SiO4:Eu2+
Abstract
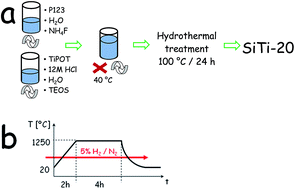
The Sr2Si0.95Ti0.05O4:Eu2+ phosphor was synthesized using titanium modified silica SBA-15 or titania as titanium precursors via a solid state synthesis method. The phase composition of samples was investigated by powder XRD technique. The SEM pictures enabled study of the influence of Ti4+ precursors on the materials' morphology. The results of versatile optical characterization pointed out that titanium incorporation causes the extending of the persistent luminescence time and does not change the basic spectral properties of the parent phosphor. The persistent luminescence phenomenon results from the presence of point defects in the lattice (ion vacancies) that play a role in electron or hole traps. It was found that both the concentration and distribution of traps in Sr2Si0.95Ti0.05O4:Eu2+ depends on the form of the titanium precursor used in the synthesis. The trap parameters were characterized by thermoluminescence measurements and luminescence kinetics analysis.
 Please wait while we load your content...
Please wait while we load your content...

