
SeO2 in water: a mild and efficient promoter for deprotection of acetyl, methoxymethyl and tetrahydropyranyl ethers and sequel oxidation of methyl/methylene carbons of alpha carbonyl carbon†
Abstract
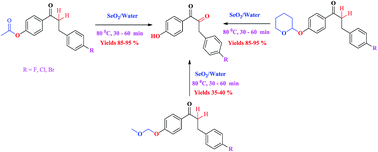
SeO2 in water provided an efficient and one-pot deprotection of acetyl, tetrahydropyranyl and methoxymethyl ethers in alcohols and phenols and sequel oxidation of methyl/methylene carbons of alpha carbonyl carbon to dicarbonyl functional groups at 80 °C in 30–60 min. Using a substrate, SeO2 in a 1 : 3 ratio, the reaction gave excellent yields (85–95%) for acetyl and tetrahydropyranyl deprotections and a moderate yield (35–40%) for methoxymethyl deprotection without affecting other functional groups.
 Please wait while we load your content...
Please wait while we load your content...

