
In vitro study of a pH-sensitive multifunctional doxorubicin–gold nanoparticle system: therapeutic effect and surface enhanced Raman scattering
Abstract
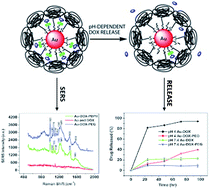
We report the development and characterization of a multifunctional drug delivery system (Au–dox–PEG) for the treatment and SERS spectroscopic detection of tumors. Doxorubicin, a therapeutic agent and a SERS tag, was chemically conjugated to gold nanoparticles via a pH-sensitive hydrazone linker, and then PEG was added to develop Au–dox–PEG. The doxorubicin occupied a maximum of 20% of the total surface area of the gold nanoparticles which resulted in colloidal stability. SERS spectra were measured for non-aggregated Au–dox–PEG using near-infrared wavelength radiation, and the doxorubicin release was time and pH dependent. Consistency in the release profile and in vitro cell viability results supports the efficacy of the Au–dox–PEG system. Thus, the development of the Au–dox–PEG multifunctional system raises exciting opportunities for the simultaneous spectroscopic detection and therapy of tumors in the future.
- This article is part of the themed collection: Surface enhanced Raman Spectroscopy: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

