DOI:
10.1039/C5RA09804A
(Paper)
RSC Adv., 2015,
5, 65133-65138
Electronic structure and thermoelectric properties of Zintl compounds A3AlSb3 (A = Ca and Sr): first-principles study†
Received
25th May 2015
, Accepted 24th July 2015
First published on 24th July 2015
Abstract
Experimentally synthesized Zn-doped Sr3AlSb3 exhibited a smaller carrier concentration than Zn-doped Ca3AlSb3, which induces a lower thermoelectric figure of merit (ZT) than Zn-doped Ca3AlSb3. We used first-principles methods and the semiclassical Boltzmann theory to study the reason for this differing thermoelectric behavior and explored the optimal carrier concentration for high ZT values via p-type and n-type doping. The covalent AlSb4 tetrahedral arrangement exhibited an important effect on the electronic structure and thermoelectric properties. p-type Ca3AlSb3 may exhibit good thermoelectric properties along its covalent AlSb4 chain due to its double band degeneracy at the valence band edge and small effective mass along its one-dimensional chain direction. Zn doping the Al site exhibited higher formation energy for Sr3AlSb3 than Ca3AlSb3, which explains the lower carrier concentration for Zn-doped Sr3AlSb3 than Zn-doped Ca3AlSb3. The double band degeneracy at the valence band edge for Ca3AlSb3 may also help to increase the carrier concentration. Sr3AlSb3 containing isolated Al2Sb6 dimers can exhibit a high thermoelectric performance via heavy p-type doping with a carrier concentration above 1 × 1020 holes per cm3. Moreover, the ZT maxima for the n-type Sr3AlSb3 can reach 0.76 with a carrier concentration of 4.5 × 1020 electrons per cm3.
I. Introduction
Thermoelectric materials can convert energy between heat and electricity and have attracted significant attention. The conversion efficiency for thermoelectric materials is defined by the dimensionless figure of merit, ZT = S2σT/κ, where S, σ, T, and κ are the Seebeck coefficient, electrical conductivity, absolute temperature, and thermal conductivity, respectively. Therefore, a promising thermoelectric material should have a large Seebeck coefficient, high electrical conductivity, and low thermal conductivity. However, S, σ, and κ are always coupled, which makes it difficult for a material to simultaneously have high S, high σ, and low κ.1 The concept of a “phonon glass electron crystal” (PGEC) is a good approach for achieving good thermoelectric performance.2 Materials with complex structures often have low intrinsic lattice thermal conductivities and may have good thermoelectric properties.3,4 Zintl compounds have emerged as a promising class of materials for thermoelectric applications due to their complex crystal structures5,6 and low intrinsic lattice thermal conductivity. For example, the lattice thermal conductivities of Ca3AlSb3, Sr3AlSb3, and Sr3GaSb3 are below 0.6 W mK−1 at 1000 K.4,7,8
Finding cheap, nontoxic thermoelectric materials with high conversion efficiencies is important.9 Ca3AlSb3 and Sr3AlSb3 are promising compounds for thermoelectric applications due to their earth abundant, nontoxic elemental composition. The transport properties for Ca3AlSb3 and Zn-doped Ca3AlSb3 were experimentally investigated,4,10 and the thermoelectric properties of Ca3AlSb3 were improved via p-type doping to obtain a maximum ZT = 0.8 at 1050 K.10 Sr3AlSb3 exhibits a maximum ZT value of 0.1 at 1000 K. Zinc doping has little effect on the carrier concentration in Sr3AlSb3 and does not improve the ZT.7 Comparing the properties for Ca3AlSb3 and Sr3AlSb3 ref. 4, 7 and 10 reveals an interesting phenomenon: Zn-doped Sr3AlSb3 exhibits a lower carrier concentration than Zn-doped Ca3AlSb3 with a maximum ZT value of approximately 0.15 at 800 K.7 Such a low carrier concentration could cause the low ZT for Sr3AlSb3. Hence, improving the low Sr3AlSb3 carrier concentration is important to improve its thermoelectric performance. Carrier concentrations always exhibit opposing effects on S, σ, and κe. Thus, optimizing the carrier concentration can increase the ZT values for Ca3AlSb3 and Sr3AlSb3. To explain these phenomenon and improve the thermoelectric properties, we calculated the electronic structures and thermoelectric properties for A3AlSb3 (A = Ca, Sr) using a first-principles method and the semiclassical Boltzmann theory. We found that p-type Ca3AlSb3 is a promising thermoelectric compounds along the chain direction. Meanwhile, n-type and p-type Sr3AlSb3 may also have good thermoelectric properties via heavy doping.
II. Computational detail
Experimental A3AlSb3 (A = Ca, Sr) structures are used as the initial bulk model and relaxed to obtain the minimum energy structure.11,12 The structure was optimized using the projector-augmented-wave (PAW) method based on density functional theory (DFT) as implemented in Vienna ab initio simulation package (VASP).13–15 The exchange-correlation function was treated with the Perdew–Burke–Ernzerhof (PBE) generalized-gradient approximation (GGA).16–18 A plane wave cutoff energy of 500 eV was used throughout. For the Brillouin zone integration, 4 × 11 × 3 and 2 × 7 × 4 Monkhorst-Pack special k-point grids were used for Ca3AlSb3 and Sr3AlSb3, respectively.
We calculated the electronic structure via the full-potential linearized augmented plane waves (FLAPW) method19 implemented in WIEN2k.20–22 The exchange-correlation potential was below the generalized gradient approximation (GGA) as parameterized by Perdew, Burke, and Ernzerhof (PBE).16 A plane wave basis cutoff of RmtKmax = 8 was used. The muffin tin radius was 2.5 a.u. for Ca, Sr, Al, and Sb. Self-consistent calculations were performed using 500 k points in the irreducible Brillouin zone for both Ca3AlSb3 and Sr3AlSb3. We obtained accurate band gaps using the modified Becke–Johnson (TB-mBJ)23 semilocal exchange potential, and the TB-mBJ method reliably predicted the band gap for sp-bonded semiconductors.24 We included the scalar-relativistic and spin–orbit coupling effects (RSO) to account for Sr due to its large atomic mass.
The thermoelectric properties were evaluated via the semiclassical Boltzmann theory25–27 as implemented in the Boltz-Trap code.28 The constant scattering time approximation was used, assuming that the scattering time determining the electrical conductivity does not vary significantly with energy on the scale of kT.
III. Results and discussions
A. Crystal structure and properties
Two structure types were formed in the A3AlSb3 compounds (A = alkaline earth metals): Ca3InP3 (no. 62 Pnma) and Ba3AlSb3 (no. 64 Cmca). The tetrahedral A3AlSb3 (A = Ca, Sr) structures determined by our simulations are shown in Fig. 1. The figure does not show the A site atoms to better show the tetrahedral AlSb4 arrangement. The Ca3AlSb3 had an anisotropic one-dimensional structure that formed a Ca3InP3 structure from infinite chains of corner-sharing tetrahedral AlSb4 linked by Ca ions.29 Sr3AlSb3 formed a Ba3AlSb3 structure type. The Al2Sb612− dimer in Sr3AlSb3 are shown in Fig. 1 and formed from two tetrahedral sharing a common edge as for Al2Cl6. Meanwhile, Sr and Sb formed a distorted NaCl-type arrangement with Al in tetrahedral holes.
 |
| | Fig. 1 The tetrahedral structure of A3AlSb3 (A = Ca, Sr) with space group Pnma, and Cmca, respectively. The blue and brown spheres represent Al and Sb atoms, respectively. | |
The optimized structure parameters and bond lengths are listed in Tables 1 and 2. The lattice constants and atomic positions of the optimized structures are close to the experimental values.11,12 The bond lengths between Al and the neighboring Sb atoms in A3AlSb3 (A = Ca, Sr) were 2.73 Å, and 2.71 Å, respectively. That is, Sr3AlSb3 may have stronger Al–Sb interactions than Ca3AlSb3. In addition, Table 1 indicates each Ca3AlSb3 primitive cell contained seven inequivalent atoms, and each Sr3AlSb3 one contained five inequivalent atoms. However, each Sr3AlSb3 primitive cell had 56 atoms in common with Sr3GaSb3, while each Ca3AlSb3 cell had only 28 atoms in common. This complex structure suggests Sr3AlSb3 should have a lower lattice thermal conductivity (approximately 0.55 W mK−1 at 1000 K) than Ca3AlSb3 (approximately 0.60 W mK−1 at 1000 K). The large cell and complex structure always induce short mean-path-lengths of heat carrying phonons.30
Table 1 Calculated lattice parameters and fractional atomic coordinated for Wyckoff sites of optimized A3AlSb3 (A = Ca, Sr)
| Lattice parameter |
Atomic type |
Wyckoff symbol |
x |
y |
z |
| Orthorhombic |
Ca1 |
4c |
0.27240 |
0.25000 |
0.27952 |
| No. 62 Pnma |
Ca2 |
4c |
0.55863 |
0.25000 |
0.38798 |
| a = 12.9637 Å |
Ca3 |
4c |
0.35002 |
0.25000 |
0.99673 |
| b = 4.5191 Å |
Al |
4c |
0.56762 |
0.25000 |
0.79666 |
| c = 14.3421 Å |
Sb1 |
4c |
0.61368 |
0.25000 |
0.60928 |
| Sb2 |
4c |
0.75644 |
0.25000 |
0.88129 |
| Sb3 |
4c |
0.04036 |
0.25000 |
0.35077 |
| Orthorhombic |
Sr1 |
8f |
0.17600 |
0.30314 |
0.12913 |
| No. 64 Cmca |
Sr2 |
16g |
0.00000 |
0.19028 |
0.35367 |
| a = 20.6293 Å |
Al |
8d |
0.08617 |
0.00000 |
0.00000 |
| b = 6.9666 Å |
Sb1 |
8f |
0.33948 |
0.28833 |
0.12283 |
| c = 13.6355 Å |
Sb2 |
16g |
0.00000 |
0.21262 |
0.10750 |
Table 2 Calculated bond distances (in Å) of optimized A3AlSb3 (A = Ca, Sr)
| Atomic type |
Nearest neighbor table |
| Ca1 |
Sb1 3.14 |
Sb3 3.18 |
Sb2 3.25 |
Al 3.26 |
|
|
| Ca2 |
Sb1 3.18 |
Sb1 3.25 |
Sb2 3.30 |
|
|
|
| Ca3 |
Sb2 3.17 |
Sb3 3.30 |
Sb3 3.39 |
Sb1 3.42 |
|
|
| Al |
Sb2 2.73 |
Sb1 2.75 |
Sb3 2.77 |
Ca1 3.26 |
|
|
| Sb1 |
Al 2.75 |
Ca1 3.14 |
Ca2 3.18 |
Ca2 3.25 |
|
|
| Sb2 |
Al 2.73 |
Ca3 3.17 |
Ca1 3.25 |
Ca2 3.30 |
|
|
| Sb3 |
Al 2.77 |
Ca1 3.18 |
Ca3 3.30 |
Ca3 3.39 |
|
|
| Sr1 |
Al 3.32 |
Sb1 3.38 |
Sb1 3.40 |
Sb1 3.51 |
Sb1 3.60 |
Sb2 3.70 |
| Sr2 |
Sb2 3.36 |
Sb2 3.37 |
Sb1 3.40 |
Sb1 3.40 |
Al 3.43 |
Al 3.43 |
| Al |
Sb1 2.71 |
Sb1 2.71 |
Sb2 2.74 |
Sb2 2.74 |
Sr1 3.32 |
Sr1 3.72 |
| Sb1 |
Al 2.71 |
Sr1 3.38 |
Sr1 3.40 |
Sr2 3.40 |
Sr1 3.40 |
|
| Sb2 |
Al 2.74 |
Al 2.74 |
Sr2 3.36 |
Sr2 3.37 |
|
|
Previous experimental work by Zevalkink et al. found Zn-doped Sr3AlSb3 possessed a lower carrier concentration than Zn-doped Ca3AlSb3.4,7,10 To explain the low carrier concentration in Sr3AlSb3, we calculated the formation energies (Ef) for A3AlSb3 (A = Ca, Sr) using a Y atom to replace X (X = Ca, Sr, Al, Sb, Si, Ge, or Sn) in the 1 × 4 × 1 and 1 × 2 × 1 supercell. The formation energy was calculated using
| | |
Ef = Edoped − Ebulk − EX + EY,
| (1) |
where,
Edoped and
Ebulk are the total energies for the supercell containing the substitutional dopant Y and the same bulk A
3AlSb
3 supercell, respectively.
EY and
EX are the doping and host atom energies in the bulk phase. The calculated formation energies are shown in
Table 3. First, Zn more easily replaces Al than Sb. Second, Zn more easily dopes in the Al site in Ca
3AlSb
3 than Sr
3AlSb
3, possibly due to the stronger bond between Al and Sb in Sr
3AlSb
3. Therefore, Zn-doped Sr
3AlSb
3 has a lower carrier concentration than Zn-doped Ca
3AlSb
3, as reported in
ref. 4, 7 and 10. Third, the formation energy for doping the A site is lower for K than Na, which was probably due to K exhibiting a stronger activity than Na. Lastly, the A (A = Ca, Sr) site atom is easier to replace in Ca
3AlSb
3 than Sr
3AlSb
3. n-type doping for Ca
3AlSb
3 and Sr
3AlSb
3 has never been performed experimentally. It is interesting to explore the possibility of n-type doping in these two compounds. Thus, we also calculated the formation energies of Si, Ge, and Sn on Al sites. As shown in
Table 3, doping of Sn on Al sites is most easily performed in Ca
3AlSb
3, and doping of Ge in Al site is most easily performed in Sr
3AlSb
3 among Si, Ge, and Sn elements. The calculated formation energies shows that doping of Si, Ge, and Sn on Al site are more difficult than that of Zn on Al site.
Table 3 Calculated formation energies (in eV) of A3AlSb3 (A = Ca, Sr) by using Y atoms to replace X atoms
| |
X = A, Y = Na |
X = A, Y = K |
X = Al, Y = Zn |
X = Sb, Y = Zn |
| Ca3AlSb3 |
1.23 |
0.77 |
0.06 |
2.1 |
| Sr3AlSb3 |
1.67 |
1.63 |
0.24 |
2.3 |
| |
X = Al, Y = Si |
X = Al, Y = Ge |
X = Al, Y = Sn |
| Ca3AlSb3 |
1.13 |
0.79 |
0.68 |
| Sr3AlSb3 |
1.05 |
0.8 |
0.9 |
B. Transport properties
To optimize the carrier concentration and increase the thermoelectric performance for A3AlSb3 (A = Ca, Sr), we calculated the A3AlSb3 (A = Ca, Sr) transport coefficients for carrier concentrations from 1 × 1018 to 1 × 1022 cm−3 using the semiclassical Boltzmann theory without considering the special dopant type. Fig. 2 shows calculated transport coefficient results for n-type and p-type A3AlSb3 (A = Ca, Sr) at 850 K. Fig. 2(a) shows that the Seebeck coefficients increase and then decrease with increasing carrier concentration. This phenomenon for the Seebeck coefficient at high carrier concentrations can be explained using eqn (2). For degenerate semiconductors, the Seebeck coefficient is given by| |
 | (2) |
where kB is the Boltzmann constant, m*DOS is the density of states effective mass, T is the temperature, and n is the carrier concentration. The Seebeck coefficients for Sr3AlSb3 are larger at low doping concentrations than for Ca3AlSb3 mainly due to the larger band gap (Smax = Eg/(2eTmax)). The Seebeck coefficients for p-doped A3AlSb3 are slightly larger than when n-doped due to the slightly larger density of states effective mass. Fig. 2(b) shows the electrical conductivities of A3AlSb3 (A = Ca, Sr) as a function of carrier concentration. This figure shows that the electrical conductivities increase with increasing carrier concentration regardless the doping type, which is indicative of thermal carrier activation across the band gap and supports the electrical conductivity being proportional to the carrier concentration (σ = neη). The electrical conductivity for n-type Ca3AlSb3 first increased and then decreased. The σ is much larger for Ca3AlSb3 than Sr3AlSb3 due to its smaller band mass m*b and one-dimensional chain structure. The σ of A3AlSb3 is larger for n-doping than p-doping mainly due to the smaller band mass at the bottom of conduction bands than that at the top of valence bands. Fig. 2(c) shows the ZT as a function of carrier concentration. We find the ZT of Sr3AlSb3 was larger for n-type doping than p-type doping due to the higher electrical conductivity. The maximum ZT for n-type Sr3AlSb3 reached 0.76, which corresponds to a carrier concentration of 4.5 × 1020 e cm−3 and is almost equal to the maximum experimental ZT for p-type Ca3AlSb3 (0.8 at 1050 K).
 |
| | Fig. 2 Calculated transport properties of A3AlSb3 (A = Ca, Sr): (a) Seebeck coefficients; (b) electrical conductivities; (c) ZeT. | |
It is well known that materials with good thermoelectric properties often have a high anisotropy in their crystal structures.31 We determined the best transport direction for A3AlSb3 (A = Ca and Sr) using the calculated anisotropic transport coefficients for p-type and n-type A3AlSb3 (A = Ca, Sr) as a function of the carrier concentration from 1 × 1018 to 1 × 1021 cm−3 along the x, y, and z directions at 850 K, without considering special dopants as shown in Fig. 3. This figure shows the anisotropy of the thermoelectric properties was stronger for Ca3AlSb3 than Sr3AlSb3 due to its band mass anisotropy in different directions and one-dimensional chain in Ca3AlSb3. For Ca3AlSb3, both the Seebeck coefficient and σ/τ were higher along the y direction than the x and z directions. This phenomenon is consistent with a one-dimensional chain feature along the y direction in Ca3AlSb3
 |
| | Fig. 3 Calculated anisotropy of thermoelectric properties of Ca3AlSb3 (left) and Sr3AlSb3 (right) as a function of carrier concentration from 1 × 1018 to 1 × 1021 cm−3 along the x, y, and z directions at 850 K. | |
C. Electronic structure
The basic bonding features are reflected in the calculated density of states (DOS) for A3AlSb3 (A = Ca, Sr) for energies ranging from −2 eV to 2 eV, as shown in Fig. 4. The Ca or Sr cations mainly contribute to the A3AlSb3 conduction bands, the top valence bands are dominated by the Sb states, and the Al states appear in both the valence and conduction band edges. The valence band maxima (VBM) is dominated by Sb p states, likely due to non-bonding Sb lone-pairs. The conduction band minimum (CBM) is dominated by the s and d orbitals. As shown in Fig. 4(c) and (h), the Al s states are dominant at the bottom of conduction band. Fig. 4(d) and (i) show the Sb p states contributions exceed the A p states and Al p states. For Ca3AlSb3, the Sb p states differ along the x, y, and z directions. In contrast, the Sb p states for Sr3AlSb3 were almost identical in the three directions, which indicates a strong hybridization between the Sb px, py and pz.
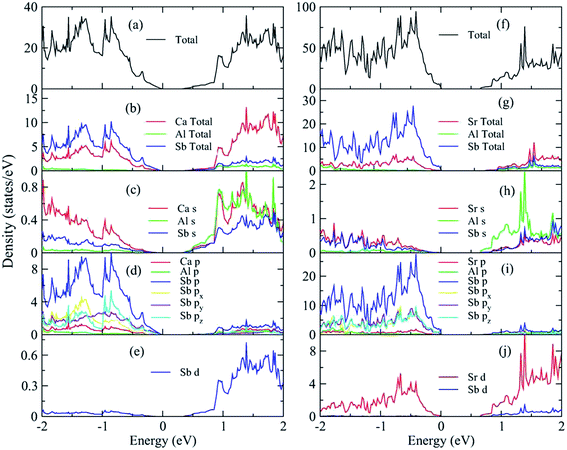 |
| | Fig. 4 Calculated total and partial DOS for A3AlSb3 (A = Ca, Sr). The Fermi level is at zero. | |
The calculated band structures for A3AlSb3 (A = Ca, Sr) are depicted in the high symmetry directions in Fig. 5, and the corresponding special k points are shown in Fig. S1, ESI.† The direct band gap is located at the Γ point for both compounds. There are two bands at the band edge for the upper valence bands. For Ca3AlSb3, the two bands at Γ and X are degenerate with a large dispersion. However, the two bands for Sr3AlSb3 are not strictly degenerate and have a relative small dispersion. Moreover, the two bands at X split in Sr3AlSb3. The difference in the band structures for these two compounds arises from their different polyanionic tetrahedral arrangements. For Ca3AlSb3, the edge-sharing anionic tetrahedra form one-dimensional chains. However, for Sr3AlSb3, the two edge-sharing tetrahedral form isolated Al2Sb6 dimers. The one-dimensional covalent chain feature enhance the covalency degree in the chain direction with a small induced band effective mass in Ca3AlSb3. Consequently, Ca3AlSb3 exhibits a larger band dispersion in the upper valence bands and lower conduction bands than Sr3AlSb3. The carrier mobility determined by A. Zevalkink4,7 confirmed this prediction.
 |
| | Fig. 5 Calculated band structures of Ca3AlSb3 (left panel) and Sr3AlSbs (right panel). The high symmetry k points Γ, X, Y, Z, S, U, and R represent the points (0, 0, 0), (0.5, 0, 0), (0, 0.5, 0), (0, 0, 0.5), (0, 0.5, 0.5), (0.5, 0.5, 0), and (0.5, 0.5, 0.5), respectively. | |
The carrier effective mass always exhibits opposing effect on the Seebeck coefficient and carrier mobility. A heavy density-of-states effective mass (m*DOS) yields a high Seebeck coefficient, and a high mobility always requires a light inertial effective mass (m*I) in the transport direction. The m*DOS, which is determined by the band mass and band degeneracy (Nv), can be defined as
| | |
m*DOS = (m*xm*ym*z)1/3Nv2/3,
| (3) |
where
m*x,
m*y, and
m*z are the band mass components along the three perpendicular directions
x,
y, and
z, respectively.
Fig. 5 and
Table 4 show that A
3AlSb
3 band gaps are 0.71 eV and 0.89 eV, which are close to the experimental values of 0.65 eV and 0.70 eV, respectively. The
m*Γ–X,
m*Γ–Y, and
m*Γ–Z for the p-type Ca
3AlSb
3 are lower than for the p-type Sr
3AlSb
3, which corresponds to the higher carrier mobility in the p-type Ca
3AlSb
3. However, the
m*DOS for the p-type Ca
3AlSb
3 were almost equal to those for p-type Sr
3AlSb
3 due to the double band degeneracy (
Nv = 2) at the valence band edge for Ca
3AlSb
3. These results are consistent with the experimental Seebeck coefficient being slightly smaller for p-type Ca
3AlSb
3 than p-type Sr
3AlSb
3. For thermoelectric materials with carriers scattered mainly by acoustic phonons, a light band mass induces a high carrier mobility and improves the thermoelectric performance, which exceeds the detrimental effects on Seebeck coefficient due to the low effective mass.
32 Thus, Ca
3AlSb
3 is a promising thermoelectric material.
Table 4 Calculated electronic properties of A3AlSb3: band gap (Eg in eV); band degeneracy (Nv); the band mass along three perpendicular directions (m* in me); and density of states effective mass (m*DOS in me)
| |
Eg (eV) |
Nv |
m*Γ–X |
m*Γ–Y |
m*Γ–Z |
m*DOS |
| n-type Ca3AlSb3 |
0.71 |
1 |
0.67 |
0.12 |
0.91 |
0.42 |
| p-type Ca3AlSb3 |
0.71 |
2 |
0.67 |
0.57 |
0.62 |
0.98 |
| n-type Sr3AlSb3 |
0.85 |
1 |
0.51 |
0.36 |
0.78 |
0.52 |
| p-type Sr3AlSb3 |
0.85 |
1 |
0.87 |
0.85 |
1.31 |
0.99 |
The optimal electronic performance for a thermoelectric semiconductor depends on its weighted mobility,2,33,34 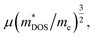 where μ and me are the carrier mobility and electron mass, respectively. Charge carriers predominantly scattered by acoustic phonons (as occurs in most good thermoelectric materials) are expected to have
where μ and me are the carrier mobility and electron mass, respectively. Charge carriers predominantly scattered by acoustic phonons (as occurs in most good thermoelectric materials) are expected to have  . Therefore, increasing the band mass should decrease the mobility. However, multiply degenerate valleys produce large m*DOS without explicitly reducing μ. A large valley degeneracy of Nv can increase m*DOS to yield a large Seebeck coefficient. Thus, a large valley degeneracy is helpful for thermoelectric materials.35,36 A thermoelectric material with a large Nv may simultaneously have light bands (small mi) and large m*DOS. The Nv for Ca3AlSb3 and Sr3AlSb3 are 2 and 1, respectively. The contribution from other bands in heavily doped Sr3AlSb3 with maxima at approximately −0.1 eV could potentially lead to an Nv of 2, which corresponds to a carrier concentration of 1 × 1020 cm−3. Thus, a heavily p-type doped Sr3AlSb3 may exhibit better thermoelectric properties at carrier concentrations above 1 × 1020 cm−3.
. Therefore, increasing the band mass should decrease the mobility. However, multiply degenerate valleys produce large m*DOS without explicitly reducing μ. A large valley degeneracy of Nv can increase m*DOS to yield a large Seebeck coefficient. Thus, a large valley degeneracy is helpful for thermoelectric materials.35,36 A thermoelectric material with a large Nv may simultaneously have light bands (small mi) and large m*DOS. The Nv for Ca3AlSb3 and Sr3AlSb3 are 2 and 1, respectively. The contribution from other bands in heavily doped Sr3AlSb3 with maxima at approximately −0.1 eV could potentially lead to an Nv of 2, which corresponds to a carrier concentration of 1 × 1020 cm−3. Thus, a heavily p-type doped Sr3AlSb3 may exhibit better thermoelectric properties at carrier concentrations above 1 × 1020 cm−3.
IV. Conclusion
In summary, the structural, thermoelectric, and electronic properties for A3AlSb3 (A = Ca, Sr) were studied using the first-principles calculations and semiclassical Boltzmann theory. We found the different thermoelectric properties for Ca3AlSb3 and Sr3AlSb3 were mainly derived from their different AlSb4 tetrahedral arrangement. The one-dimensional AlSb4 chain in Ca3AlSb3 yields a stronger anisotropy in the thermoelectric properties than for Sr3AlSb3 with isolated Al3Sb6 dimers. On the one hand, the one-dimensional chain structure for Ca3AlSb3 exhibited high electrical conductivities along the chain direction. On the other hand, the double band degeneracy at the valence band edge creates a large Seebeck coefficient for p-type Ca3AlSb3. Consequently, Ca3AlSb3 may exhibit good thermoelectric properties along the chain direction, which requires further research experiment. The formation energies calculated for Zn doped at Al sites revealed the origin of lower carrier concentrations of Zn-doped Sr3AlSb3 than Zn-doped Ca3AlSb3 as reported by experimental workers. The transport properties for Sr3AlSb3 indicate the highest achievable ZT for n-type Sr3AlSb3 was 0.76, which corresponds to a carrier concentration of 4.5 × 1020 cm−3.
Acknowledgements
This research was sponsored by the National Natural Science Foundation of China (No. 51371076 and U1204112), the Program for Innovative Research Team (in Science and Technology) in University of Henan Province (No. 13IRTSTHN017).
References
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105 CrossRef CAS PubMed
 .
. - G. A. Slack, CRC Handbook of Thermoelectrics, ed. D. M. Rowe, CRC, Boca Raton, FL, 1995, pp. 406–440 Search PubMed .
- Y. L. Yan and Y. X. Wang, J. Mater. Chem., 2011, 21, 12497–12502 RSC .
- A. Zevalkink, E. S. Toberer, W. G. Zeier, E. Flaage-Larsen and G. J. Snyder, Energy Environ. Sci., 2011, 4, 510 CAS .
- S. M. Kauzlarich, S. R. Brown and G. J. Snyder, Dalton Trans., 2007, 2099 RSC .
- E. S. Toberer, A. F. May and G. J. Snyder, Chem. Mater., 2010, 22, 624 CrossRef CAS .
- A. Zevalkink, G. Pomrehn, Y. Takagiwa, J. Swallow and G. J. Snyder, ChemSusChem, 2013, 6, 2316 CrossRef CAS PubMed .
- Q. F. Shi, Y. L. Yan and Y. X. Wang, Appl. Phys. Lett., 2014, 104, 012104 CrossRef PubMed .
- L. E. Bell, Science, 2008, 321, 1457 CrossRef CAS PubMed .
- W. G. Zeier, A. Zevalkink and G. J. Snyder, J. Mater. Chem., 2010, 22, 624 CrossRef .
- G. Cordier, M. Stelter and H. J. Schaefer, J. Less-Common Met., 1984, 98, 285 CrossRef CAS .
- G. Cordier, G. Savelsberg and H. Z. Schaefer, Z. Naturforsch., B: J. Chem. Sci., 1982, 37, 975 Search PubMed .
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS .
- G. Dresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245 CrossRef .
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS .
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS .
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef .
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS .
- D. Singh, Planewaves, Pseudopentials, and the LAPW Method, Kluwer Academic, Boston, 1994 Search PubMed .
- D. D. Koelling and B. N. Harmon, J. Phys. C: Solid State Phys., 1977, 10, 3107 CrossRef CAS PubMed .
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, 864 CrossRef .
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka and J. Luitz, WIEN2K, An Augmented Plane Wave+Local Orbitals Program for Calculating Crystal Properties, Vienna University of Technology, Vienna, Austria, 2001 Search PubMed .
- F. Tran and P. Blaha, Phys. Rev. Lett., 2009, 102, 226401 CrossRef .
- D. Koller, F. Tran and P. Blaha, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195134 CrossRef .
- J. M. Ziman, Electrons and Phonons, Oxford University Press, New York, 2001 Search PubMed .
- W. Jone and N. H. March, Theoretical Solid State Physics, Courier Dover, New York, 1985 Search PubMed .
- G. K. H. Madsen, K. Schwarz, P. Blaha and D. J. Singh, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 125212 CrossRef .
- T. Thonhauser, T. J. Scheidemantel, J. O. Sofo, J. V. Badding and G. D. Mahan, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 085201 CrossRef .
- G. Cordier, H. Schaefer and M. Stelter, Z. Naturforsch., B: J. Chem. Sci., 1984, 37, 975 Search PubMed .
- J. R. Sootsman, D. Y. Chung and M. G. Kanatzidis, Angew. Chem., Int. Ed., 2009, 48, 8616 CrossRef CAS PubMed .
- T. J. Scheidemantel, C. Ambrosch-Draxl, T. Thonhauser, J. V. Badding and J. O. Sofo, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 125210 CrossRef .
- Y. Pei, A. D. Lalonde, H. Wang and G. J. Snyder, Energy Environ. Sci., 2012, 5, 7963 CAS .
- G. D. Mahan, Solid State Physics, Academic Press Inc, San Diego, 1998, vol. 51, p. 81 Search PubMed .
- H. J. Goldsmid, Thermoelectric Refrigeration, Plenum, 1964 Search PubMed .
- F. J. DiSalvo, Science, 1999, 285, 703 CrossRef CAS .
- Y. Pei, X. Shi, A. LaLonde, H. Wang, L. Chen and G. J. Snyder, Nature, 2011, 473, 66 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09804a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






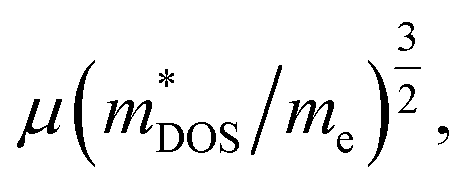 where μ and me are the carrier mobility and electron mass, respectively. Charge carriers predominantly scattered by acoustic phonons (as occurs in most good thermoelectric materials) are expected to have
where μ and me are the carrier mobility and electron mass, respectively. Charge carriers predominantly scattered by acoustic phonons (as occurs in most good thermoelectric materials) are expected to have  . Therefore, increasing the band mass should decrease the mobility. However, multiply degenerate valleys produce large m*DOS without explicitly reducing μ. A large valley degeneracy of Nv can increase m*DOS to yield a large Seebeck coefficient. Thus, a large valley degeneracy is helpful for thermoelectric materials.35,36 A thermoelectric material with a large Nv may simultaneously have light bands (small mi) and large m*DOS. The Nv for Ca3AlSb3 and Sr3AlSb3 are 2 and 1, respectively. The contribution from other bands in heavily doped Sr3AlSb3 with maxima at approximately −0.1 eV could potentially lead to an Nv of 2, which corresponds to a carrier concentration of 1 × 1020 cm−3. Thus, a heavily p-type doped Sr3AlSb3 may exhibit better thermoelectric properties at carrier concentrations above 1 × 1020 cm−3.
. Therefore, increasing the band mass should decrease the mobility. However, multiply degenerate valleys produce large m*DOS without explicitly reducing μ. A large valley degeneracy of Nv can increase m*DOS to yield a large Seebeck coefficient. Thus, a large valley degeneracy is helpful for thermoelectric materials.35,36 A thermoelectric material with a large Nv may simultaneously have light bands (small mi) and large m*DOS. The Nv for Ca3AlSb3 and Sr3AlSb3 are 2 and 1, respectively. The contribution from other bands in heavily doped Sr3AlSb3 with maxima at approximately −0.1 eV could potentially lead to an Nv of 2, which corresponds to a carrier concentration of 1 × 1020 cm−3. Thus, a heavily p-type doped Sr3AlSb3 may exhibit better thermoelectric properties at carrier concentrations above 1 × 1020 cm−3.