
An efficient one pot ipso-nitration: structural transformation of a dipeptide by N-terminus modification†
Rajib Sarkar,
Krishnendu Maji and
Debasish Haldar*
Department, of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, West Bengal 741246, India. E-mail: deba_h76@yahoo.com; deba_h76@iiserkol.ac.in; Fax: +913325873020; Tel: +913325873119
First published on 3rd July 2015
Abstract
The one pot ipso-nitration of Boc-capped rigid dipeptide Boc-Maba-Aba-OMe 1 gave poor yields under excess NaNO2 in dilute mineral acidic medium at 0 °C. The yield is improved with the use of a catalytic amount of CuSO4. The N-terminus modification leads to structural transformation of the dipeptide from anti parallel to parallel β-sheet. We have also explored the reverse reaction by one pot reduction of nitro group to amine and Boc protection.
Introduction
Structural transformation-the large scale transfer of secondary structure of a protein or peptide to another structure is very important for many human diseases including Alzheimer's disease.1 Hence the modulation of the peptide or protein secondary structures may have potential for inhibition and treatment of these misfolding related diseases.2 One popular method is the modulation of a self-assembled structure as a reflection of organic small molecule–peptide interactions.3 Wang and co-workers have introduced bipyridine and terpyridine to modulate the self-assembly behavior of a critical amyloid peptide fragment KLVFF.4 Another method is the modulation of the self-assembled structure as a reflection of the minor chemical modification of the related protein or peptide.5 In this context, protective groups6 have a significant effect on the structure, function as well as the self-assembly propensities of the peptides.7 The Boc protecting group increases the crystallinity as well as hydrophobicity and introduces an additional hydrogen bond donor site within a peptide.8 On the other hand, the Fmoc protecting group increases the gelatinous and fluorescence properties.9 Cbz, Troc, Alloc etc. N-terminus protecting groups are also very popular because of their characteristic features.Aromatic nitro compounds are widely used in the preparation of many dyes, plastics, perfumes, explosives, and pharmaceuticals.10 Moreover, nitro-aryl moieties play key roles in the physical and chemical properties and self-assembly process in peptide chemistry.11 In general, in industry aromatic nitro compounds are produced by electrophilic nitration of arenes with HNO3 in drastic acidic conditions. Under these drastic conditions functional group tolerance, over-nitration and regioselectivity can be problematic. Hence, ipso-nitration is very important for functional group modification. Prakash and Olah et al. have reported ipso-nitration of aryl boronic acids by applying AgNO3 or NH4NO3.12 Recently, Buchwald and co-workers have developed a general and efficient procedure for the palladium-catalyzed ipso-nitration of aryl chlorides and sulfonates using NaNO2 as nitro group source.13
Intriguing by the previous knowledge, we wanted to investigate the structural transformation of peptides with functional group modification. Herein we present influence of the ipso-nitration reaction on secondary structure of a dipeptide. The dipeptide 1 contains Boc protected m-aminobenzoic acid and α-aminobutyric acid. The ipso-nitration reaction replace the Boc protected NH group by a nitro group. But the rigid dipeptide Boc-Maba-Aba-OMe 1 gave poor yields under excess NaNO2 in dilute mineral acidic medium at 0 °C. However, the yield has significantly improved with the addition of catalytic amount of CuSO4. From UV/Vis and FT-IR spectroscopic studies the peptides 1 and 2 have similar self-assembly pattern. Single crystal X-ray diffraction studies exhibit that the dipeptide 1 adopts intermolecular hydrogen bonded anti parallel sheet-like structure. But in solid state peptide 2 adopts a parallel sheet-like structure in higher order assembly. Finally we have explored the possibility of reverse reaction by one pot reduction of nitro group to amine and Boc protection. The reduction by H2 and Pd/C in dioxane solution of peptide 2 led to formation of the amine and further addition of (Boc)2O and Et3N provides the dipeptide 1 in quantitative yield.
Results and discussion
The dipeptides were designed with assumption that the rigid m-aminobenzoic acid will retain the conformational preferences. The one pot ipso-nitration leads to the replacement of BocNH group by a nitro group, but rest of the primary structures are same (Scheme 1). Similarly, the one pot reduction of nitro group to amine and Boc protection leads to Boc protected peptides, but rest of the primary structures are same. In this context the folding and self-assembly of the peptides may be identical. Target dipeptide 1 was synthesized by coupling Boc protected m-aminobenzoic acid with α-aminobutyric acid methyl ester. The ipso-nitration of peptide 1 provides peptide 2. The synthesized compounds were purified and characterized by 1H-NMR, 13C-NMR, FT-IR and mass spectrometry (MS) analysis. | ||
| Scheme 1 The schematic presentation of ipso-nitration reaction of peptide 1. | ||
Inspired from Sandmeyer reaction,14 the ipso-nitration tried between Boc-capped peptide 1 with excess NaNO2 in dilute mineral acidic medium at 0 °C. The assumption was that in presence of aqueous acid Boc group will be removed and NaNO2 will provide nitrous acid which further diazotize the Boc free amine. Finally, the diazonium group will be substitute by nitrous acid14 and deprotonation of the resulting compound will produce the desired nitro compound. But peptide 1 has solubility problem in aqueous acid. The addition of trifluoroacetic acid makes the peptide soluble. The use of dilute sulphuric acid confirms that the counter anion will not be the competitor of nitrous acid in diazonium reaction (Scheme 2). The yield was 40%. However, the yield was improved to 60% by the addition of catalytic amount of CuSO4. The hexa-aqua Cu2+ complex stabilized the unstable nitrous acid in acidic medium and improved the yield. Optimization studies of catalytic amount of copper, reaction time and temperature have been reported in the ESI.†
 | ||
| Scheme 2 The proposed mechanism of ipso-nitration reaction. | ||
To investigate the impact of ipso-nitration reaction on the structure and self-assembly behaviour of dipeptides 1 and 2, different spectroscopic techniques have been used. The typical UV/Vis absorption spectra of dipeptide 1 in methanol (0.023 mM) show an absorption band at 220 nm for π to π* transition (Fig. 1a). However, increasing the concentration of dipeptide 1 induces stacking interactions between the molecules. The absorption spectra of dipeptide 2 in methanol also show an absorption band at 215 nm and intensity increases with increasing concentration (Fig. 1b). These results show that the dipeptides 1 and 2 have similar aggregation pattern.
 | ||
| Fig. 1 Concentration dependent UV/Vis spectra of (a) dipeptide 1 and (b) dipeptide 2 showing similar aggregation pattern in methanol. | ||
FT-IR analysis is an excellent method to investigate the structure and self-assembly of the reported dipeptides. In FT-IR, the region of 3500–3200 cm−1 is responsible for the N–H stretching vibrations.15 The range 1800–1500 cm−1 is assigned for the stretching band of amide I and the bending peak of amide II and ester groups. The FT-IR spectra of dipeptide 1 (Fig. 2a) exhibits N–H stretching frequency at 3325 cm−1 for hydrogen bonded N–H. The characteristic IR absorption band at 1639 and 1539 cm−1 suggest that the peptides have spectra typical of β-sheet structure.16 The peak around 1732 cm−1 is responsible for the non hydrogen bonded ester carbonyls. The peptide 2 exhibits peak at 3328 cm−1 for N–H stretching frequency. The amide I and II have appeared at 1628 and 1545 cm−1 (Fig. 2b). The peak around 1739 cm−1 is responsible for the non hydrogen bonded ester carbonyls. These results indicate that the dipeptides 1 and 2 have sheet-like structures.
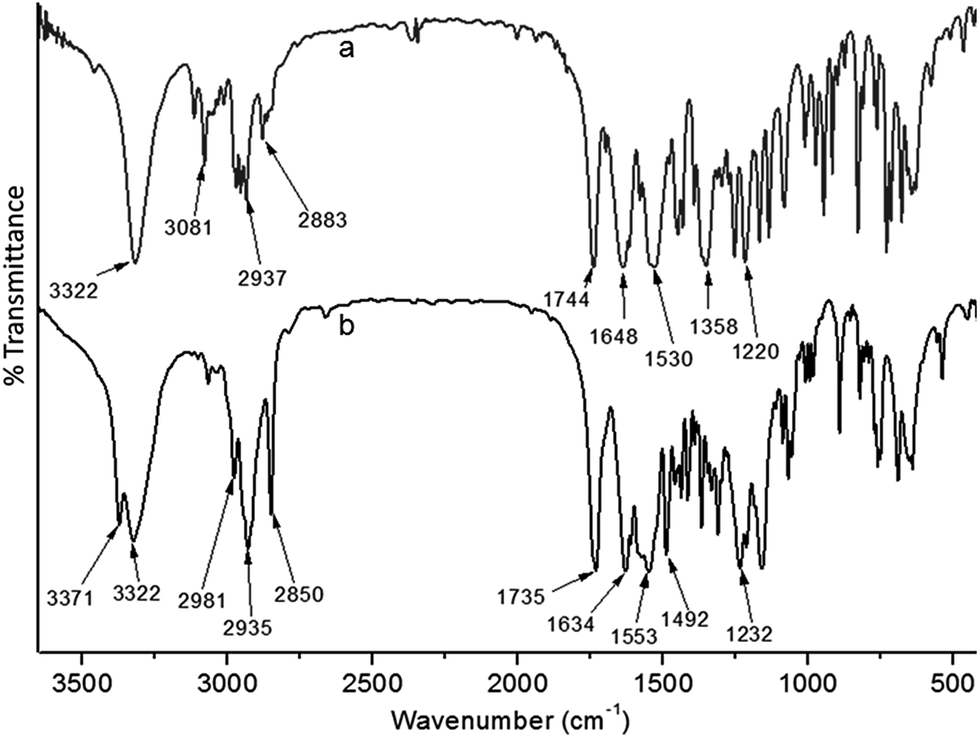 | ||
| Fig. 2 Solid state FT-IR spectra of dipeptides (a) 1 and (b) 2. | ||
Clear evidence of structural transformation has confirmed by X-ray crystallography.17 Colourless monoclinic crystals of peptide 1 and orthorhombic crystals of peptide 2 suitable for X-ray diffraction studies were obtained from an ethyl acetate–hexane solution by slow evaporation. From the crystal structure of peptide 1, due to internal rigidity, the peptide adopts a kink-like structure (ESI Fig. S2†). Dipeptide 2 crystallizes with one molecule in the asymmetric unit (ESI Fig. S3†). The solid state structure of dipeptide 2 shows that the peptide adopts kink like structure (Fig. 3b). The torsion angles around the rigid benzene ring and the Aba residues appears to play a critical role in dictating the overall structural features of dipeptides 1 and 2 (Table 1). Two molecules of peptide 1 are inter-linked through intermolecular hydrogen bonds and thereby form an anti parallel dimer along the crystallographic c-axis (Fig. 3). The dimer is also stabilized by π–π interaction (C–C distance 4.60 Å). The crystal structure further reveals that the dimeric unit of peptide 1 further self-assembled through intermolecular hydrogen bonds and stabilize the supramolecular anti parallel sheet-like structure in higher order packing along the crystallographic c and b directions (Fig. 3). But for peptide 2, the individual subunits are stacked on top of another by intermolecular H-bonding interaction between neighbouring molecules and generate a supramolecular parallel sheet-like structure (Fig. 4) about crystallographic b direction. The intermolecular hydrogen bonding parameters of peptide 1 and 2 are listed in Table 2.
 | ||
| Fig. 3 The solid state structures of dipeptide 1 showing hydrogen bonded anti parallel sheet. The intermolecular hydrogen bonds are presented as black dotted line. | ||
| Peptide 1 | Peptide 2 | ||||
|---|---|---|---|---|---|
| C5–N1–C6–C11 | −176.6 | ϕ1 | O2–N1–C7–C6 | −178.4 | ϕ1 |
| C11–C12–C10–N2 | −170.2 | ψ1 | C6–C5–C4–N2 | 144.6 | ψ1 |
| C12–N2–C13–C16 | 64.3 | ϕ2 | C4–N2–C3–C2 | 48.0 | ϕ3 |
| N2–C13–C16–O5 | −46.5 | ψ2 | N2–C3–C2-O5 | 45.5 | ψ3 |
 | ||
| Fig. 4 Parallel sheet-like packing of dipeptide 2. The intermolecular hydrogen bonds are presented as black dotted line. | ||
| D-H⋯A | D⋯H (Å) | H⋯A (Å) | D⋯A (Å) | D-H⋯A (°) | |
|---|---|---|---|---|---|
| a Symmetry equivalent a = x, 1/2 − y, −1/2 + z, b = x, 1/2 − y, 1/2 + z, c = x, 1 + y, z. | |||||
| 1 | N2–H8⋯O3a | 0.86 | 2.09 | 2.931(3) | 167 |
| N1–H17⋯O4b | 0.86 | 2.12 | 2.943(3) | 161 | |
| 2 | N2–H4⋯O3c | 0.86 | 2.06 | 2.847(6) | 152 |
We have explored the ipso nitration reaction not only on Boc-Maba-Aba-OMe 1 but also on ortho and para isomers Boc-Oaba-Aba-OMe 3 and Boc-Paba-Aba-OMe 5 respectively. We have obtained similar result (ESI†).
Next, we explored the scope of reverse reaction i.e. the amination followed by Boc protection (Scheme 3). In a typical procedure, H2 and Pd/C in dioxane solution of peptide 2 led to reduction of the nitro group to amine. Further addition of (Boc)2O and Et3N provides the dipeptide 1 in quantitative yield.
 | ||
| Scheme 3 The schematic presentation of synthesis of peptide 1 from 2. | ||
We have also performed the one pot ipso-nitration reaction on another dipeptide Boc-Maba-Aib-OMe18 that provides the corresponding nitro compound in quantitative yield. However, the one pot reduction of the nitro group to amine followed by Boc protection generates the Boc-Maba-Aib-OMe in 86% yield.
Experimental
General
All amino acids were purchased from Sigma chemicals. HOBt (1-hydroxybenzotriazole) and DCC (dicyclohexylcarbodiimide) were purchased from SRL.Peptide synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as the eluent. Yield: 2.6 g (7.1 mM, 71%).:4) as the eluent. Yield: 0.16 g (0.6 mM, 60%).:hexane (1:3) as the eluent.
:3) as the eluent. Yield: 2.6 g (7.1 mM, 71%).:4) as the eluent. Yield: 0.16 g (0.6 mM, 60%).:hexane (1:3) as the eluent.Yield: Boc-Maba-Aba-OMe: 0.28 g (0.84 mM, 84%).
Boc-Maba-Aib-OMe: 0.29 g (0.86 mM, 86%).
NMR Experiments
All NMR studies were carried out on a Jeol 400 MHz and Brüker AVANCE 500 MHz spectrometer at 278 K. Compound concentrations were in the range 1–10 mM in CDCl3 or in DMSO-d6.FT-IR spectroscopy
All reported solid-state FT-IR spectra were obtained with a Perkin Elmer Spectrum RX1 spectrophotometer with the KBr disk technique.UV/Vis spectroscopy
UV/Vis absorption spectra were recorded on a UV/Vis spectrophotometer (Hitachi).Mass spectrometry
Mass spectra were recorded on a Q-Tof Micro YA263 high-resolution (Waters Corporation) mass spectrometer by positive-mode electrospray ionization.Single crystal X-ray diffraction study
Intensity data of peptides 1 and 2 crystals were collected with MoKα radiation using Bruker APEX-2 CCD diffractometer. Data were processed using the Bruker SAINT package and the structure solution and refinement procedures were performed using SHELX97 ESI.†Conclusions
In conclusion, the modulation of self-assembly propensities of a dipeptide containing m-aminobenzoic acid and α-aminobutyric acid has been reported. The structural transformation achieved by the one pot ipso-nitration and replacement of Boc-NH group by a nitro group. The X-ray crystallography reveals that the dipeptide 1 fabricate a dimeric unit which further self-assembles to form a supramolecular anti parallel sheet like structure. But the nitro modified 2 adopts a parallel sheet-like structure. Finally the possibility of reverse reaction by one pot reduction to amine and Boc protection has also performed. The result presented here may foster new studies on peptide and protein modifications.Acknowledgements
We acknowledge the CSIR, India, for financial assistance (Project No. 01/2507/11-EMR-II). R. Sarkar acknowledges the CSIR, India for research fellowship. K. Maji thanks IISER-Kolkata for fellowship.Notes and references
- (a) J. W. Kelly, Curr. Opin. Struct. Biol., 1996, 6, 11–17 CrossRef CAS PubMed; (b) J. C. Rochetand and P. T. Lansbury, Curr. Opin. Struct. Biol., 2000, 10, 60–68 CrossRef; (c) C. M. Dobson, Trends Biochem. Sci., 1999, 24, 329–332 CrossRef CAS PubMed; (d) S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 13363–13383 CrossRef CAS PubMed; (e) E. D. Roberson and L. Mucke, Science, 2006, 314, 781–784 CrossRef PubMed; (f) M. Goedert and M. G. Spillantini, Science, 2006, 314, 777–781 CrossRef CAS PubMed.
- (a) S. E. O'Connor and B. Imperiali, Chem. Biol., 1996, 3, 803–812 CrossRef; (b) W. F. Hawse, B. E. Gloor, C. M. Ayres, K. Kho, E. Nuter and B. M. Baker, J. Biol. Chem., 2013, 288, 24372–24381 CrossRef CAS PubMed.
- (a) D. S. Yang, C. M. Yip, T. H. J. Huang, A. Chakrabartty and P. E. Fraser, J. Biol. Chem., 1999, 274, 32970–32974 CrossRef CAS PubMed; (b) S. Alavez, M. C. Vantipalli, D. J. S. Zucker, I. M. Klang and G. J. Lithgow, Nature, 2011, 472, 226–229 CrossRef CAS PubMed; (c) C. Ramassamy, Eur. J. Pharmacol., 2006, 545, 51–64 CrossRef CAS PubMed.
- L. Niu, L. Liu, M. Xu, J. Cramer, K. V. Gothelf, M. Dong, F. Besenbacher, Q. Zeng, Y. Yang and C. Wang, Chem. Commun., 2014, 50, 8923–8926 RSC.
- (a) C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed; (b) A. Bandyopadhyay, S. V. Jadhav and H. N. Gopi, Chem. Commun., 2012, 48, 7170–7172 RSC; (c) A. Bandyopadhyay, S. M. Mali, P. Lunawat, K. M. P. Raja and H. N. Gopi, Org. Lett., 2011, 13, 4482–4485 CrossRef CAS PubMed; (d) A. Bandyopadhyay and H. N. Gopi, Org. Lett., 2012, 14, 2770–2773 CrossRef CAS PubMed.
- T. W. Greene and P. G. M. Wuts, Protective groups in organic synthesis, John Wiley and Sons inc, 1998 Search PubMed.
- S. Bera, P. Jana, S. K. Maity and D. Haldar, Cryst. Growth Des., 2014, 14, 1032–1038 CAS.
- (a) S. Bera, S. K. Maity and D. Haldar, CrystEngComm, 2014, 16, 4834–4841 RSC; (b) A. K. Das, P. P. Bose, M. G. B. Drew and A. Banerjee, Tetrahedron, 2007, 63, 7432–7442 CrossRef CAS.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, T. Tuttle and R. V. Ulijn, Chem. Commun., 2013, 49, 10587–10589 RSC.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, 2001 Search PubMed.
- S. K. Maity, P. Kumar, D. K. S. Ambast, B. Pal and D. Haldar, J. Mater. Chem., 2012, 22, 22198–22203 RSC.
- (a) G. K. S. Prakash, C. Panja, T. Mathew, V. Surampudi, N. A. Petasis and G. A. Olah, Org. Lett., 2004, 6, 2205–2207 CrossRef CAS PubMed; (b) S. Salzbrunn, J. Simon, G. K. S. Prakash, N. A. Petasis and G. A. Olah, Synlett, 2000, 10, 1485–1487 Search PubMed.
- B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 12898–12899 CrossRef CAS PubMed.
- (a) H. H. Hadgson, Chem. Rev., 1947, 40, 251–277 CrossRef; (b) S. E. Blondelle, B. Forood, A. R. Houghten and E. Peraz-Paya, Biochemistry, 1997, 36, 8393–8400 CrossRef CAS PubMed; (c) A. Adochitei and G. Drochioiu, Rev. Roum. Chim., 2011, 56, 783–791 CAS.
- V. Moretto, M. Crisma, G. M. Bonora, C. Toniolo, H. Balaram and P. Balaram, Macromolecules, 1989, 22, 2939–2944 CrossRef CAS.
- G. P. Dado and S. H. Gellman, J. Am. Chem. Soc., 1994, 116, 1054–1062 CrossRef CAS.
- Crystallographic data of dipeptide 1: C17H24N2O5, Mw = 336.38, monoclinic, space group P21/C, a = 12.8074(9), b = 14.8520(6), c = 10.0676(5) Å, α = 90°, β = 111.024°(7), γ = 90°, V = 1787.53(19) Å3, Z = 4, dm = 1.25 mg m−3, T = 100 K, R 1 0.0679 and wR2 0.1688 for 3935 data with I > 2σ(I). For dipeptide 2: C12H14N2O5, Mw = 507.53, orthorhombic, space group PCa21, a = 13.8537(16), b = 4.8506(4), c = 18.6706(15) Å, α = 90°, β = 90° γ = 90°, V = 1254.6(2) Å3, Z = 4, dm = 1.41 mg m−3, T = 100 K, R1 0.0690 and wR2 0.1945 for 2007 data with I > 2σ(I). Intensity data were collected with MoKα radiation using Bruker APEX-2 CCD diffractometer. Data were processed using the Bruker SAINT package and the structure solution and refinement procedures were performed using SHELX9719 ESI.†.
- S. K. Maity, S. Maity, P. Jana and D. Haldar, CrystEngComm, 2012, 14, 3156–3162 RSC.
- G. M. Sheldrick, SHELX 97, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of peptides, 1H NMR, 13C NMR, Figures ESI S1–S3, Fig. S4–S33. CCDC 1057300 and 1057301. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09789d |
| This journal is © The Royal Society of Chemistry 2015 |