
Tumor specific delivery with redox-triggered mesoporous silica nanoparticles inducing neovascularization suppression and vascular normalization
Lu Sunab,
Yu-Jie Liuab,
Zhen-Zhen Yangb and
Xian-Rong Qi*abc
aBeijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery System, Peking University, Beijing 100191, China
bSchool of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing 100191, China. E-mail: qixr@bjmu.edu.cn; Fax: +86 10 82801584; Tel: +86 10 82801584
cState Key Laboratory of Natural and Biomimetic Drugs, Peking University, Beijing 100191, China
First published on 12th June 2015
Abstract
RNA interference (RNAi) has great potential in cancer therapy, however, efficient cytoplasmic delivery still remains a major challenge. In this study, redox-responsive mesoporous silica nanoparticles with enlarged pores (denoted as MSN-siRNA/CrPEI) were designed by immobilizing polyethylenimine (PEI) via intermediate linkers of disulfide bonds onto the MSNs as caps for redox-responsive intracellular gene delivery. The MSN-siRNA/CrPEI with a high siRNA loading capacity (35 mg siRNA per g MSNs) could react to the specific reductive stimulation—upgraded glutathione concentration in tumor cells and release cargos through the rupture of disulfide bonds. Subsequently, MSN-siRNA/CrPEI was used to deliver VEGF siRNA for cancer therapy and the underlying mechanisms were explored. As we expected, MSN-siRNA/CrPEI could be readily internalized into cells, escaped from the endolysosomes and was distributed in the cytoplasm where siRNA mediated its function. MSN-siRNA/CrPEI showed remarkable anti-tumor efficacy by the suppression of neovascularization and vascular normalization after peritumoral application against mice with KB tumors, proved by interstitial fluid pressure (IFP) reduction, CD31 suppression and angioplerosis. Notably, siRNA combined with dexamethasone exerted a better treatment effect which is attributed to the strong capability of dexamethasone to decrease the IFP, and a lower IFP leads to an improvement in the delivery and efficacy of exogenously administered therapeutics. These results indicate that the tumor specific delivery of siRNA with redox-triggered mesoporous silica nanoparticles is a promising strategy to enhance therapeutic efficacy. Neovascularization suppression and vascular normalization may be beneficial for cancer inhibition.
Introduction
Angiogenesis is critical to the growth of human tumors and the development of metastasis.1,2 Amongst the many proangiogenic mechanisms identified, the vascular endothelial growth factor (VEGF) signaling pathway has been implicated as the key regulator of tumor neovascularization.3 Various therapeutic agents targeting the VEGF pathway have been successfully developed in the past decades. Bevacizumab as a humanized monoclonal VEGF specific antibody has antivascular effects in human rectal cancer.4 Sorafenib, sunitinib, pazopanib and vandetanib as angiogenesis inhibitors not only targeting VEGF, but also multiple tyrosine kinase receptors, have also been approved by the Food and Drug Administration for solid tumor therapy.5,6VEGF small interference RNA (siRNA) utilize RNA interference (RNAi) to block the VEGF signaling pathway in cancer therapy. It is specific and potent.7 However, the therapeutic application of siRNA still faces many challenges due to its instability, poor membrane permeability and short serum half-life. Currently, the promising approach widely used to initiate RNAi is the delivery of siRNA into the cytoplasm of target cells via delivery systems, including viral and non-viral vectors.8 Due to the unexpected safety risks from viral vectors, non-viral delivery systems dominate the research field, which are categorized into lipid based delivery systems, polycationic polymers and inorganic nanoparticles.8 Mesoporous silica nanoparticles (MSNs) have already been widely used as delivery systems for various active molecules. Silica materials with defined structures and surface properties are known to be biocompatible and have negligible cytotoxicity.9 Furthermore, MSNs are able to enhance the biocompatibility of several drug delivery systems, such as magnetic nanoparticles, biopolymers, and micelles.9,10 Most of all, the high surface area, large pore volume and controllable chemistry of MSNs allow for the loading of high concentrations into the pores.11
In many MSNs, hexagonally symmetrical MCM-41 is one of the most commonly studied materials for drug delivery.12 Traditional MCM-41, produced by the synthesis method of liquid crystal templating, has an average mesopore size of 2.5–3.7 nm,13–15 just a little bigger than the siRNA diameter (2.6 nm),16 thus limiting the amount of siRNA entering into the mesopores. It has also been shown that the larger the pores of the MSNs, the higher the quantity of cargo the MSNs may load,17 the faster the MSNs may release the cargo in the target cell18 and the better the effect of the treatment, especially for siRNA which acts in a dose-dependent manner meaning that a higher siRNA concentration results in more significant inhibition of protein expression.19 Thus, the MSNs employed here had their mesopores enlarged through a swelling agent incorporation method.
Generally, for delivery strategies the cargo within the MSNs must be retained and not be released until delivery to the target cell. Therefore, pores must be “sealed” after cargo loading for superior MSNs. Due to the intrinsic negative charge of the silica surface from the deprotonation of surface silanols, we took advantage of a positively charged polymer, polyethylenimine (PEI), which can be electrostatically attracted onto the surface of the MSNs as a supramolecular cap and retains the cargo within the MSNs. PEI not only has considerable positive charge but also possesses an intrinsic endosomal release capacity, known as the “proton sponge effect”.20 Thus, the PEI coated MSNs can not only protect siRNA in transit from the bloodstream to the intracellular compartment, but can also guarantee a quick escape from the endosomal/lysosomal “pitfall”, thus avoiding siRNA degradation in endosomes and lysosomes and achieving siRNA intracellular targeting. Here, low molecular weight PEI (2 kDa) was used to coat the MSNs as a cap as it has lower toxicity than long chain PEI (25 kDa).21
When the MSNs reach the target position, the cap should depart after intracellular uptake, resulting in the controlled release of the cargo. Various trigger mechanisms, such as sensitivity to a change in pH, temperature, enzyme or redox conditions, have been put forward.22 The large difference in reducing potential between the intracellular and extracellular milieu enables the redox-triggered destabilization mechanism to be a promising strategy.23 In body fluids (e.g. blood) and in extracellular matrices, a relatively high redox potential is maintained due to the low concentration of glutathione (GSH, approximately 2–20 μM). On the contrary, the concentration of GSH inside cells is 0.5–10 mM, maintaining a highly reducing environment.24 Furthermore, tumor tissue has at least a 4-fold higher concentration of GSH compared to normal tissue and as a result hypoxic tumor tissues are highly reducing. Disulfide bond crosslinking is generally used on account of the reducibility in the tumor intracellular milieu for the elevated concentration of GSH.25 Therefore, dithiobis(succinimidyl propionate) (DSP), as a redox sensitive moiety, was applied to induce the thiol to construct a disulfide bond so as to achieve sensitivity to the upgraded GSH in tumor cytosol. Thus, the redox-triggered MSNs possess “smart” multi-stage vesicles that can be stimulated sensitively according to GSH changes in the physiological compartments, which protects siRNA from degradation and provides for timely drug release.
Based on the above considerations, we report redox-triggered MSNs (denoted as MSN-siRNA/CrPEI) in the present study. The pores of the MSNs were enlarged to load more siRNA. Short chain PEI was utilized as a cap via disulfide linkages induced by DSP to protect siRNA in the pores (Fig. 1A). Once the siRNA loaded MSNs were internalized by tumor cells, the PEI on the surface of the MSNs can be removed quickly due to the cleavage of disulfide linkages in the presence of GSH secreted by the tumor cells. Subsequently, the loaded siRNA would be released from the MSNs in the cytoplasm and can play a role in gene silencing (Fig. 1B). As a result, significant anti-cancer efficacy of VEGF siRNA was identified through gene silencing, tumor interstitial fluid pressure reduction, CD31 suppression, angioplerosis and tumor shrinking after peritumoral application against mice with KB tumors. In addition, siRNA combined with dexamethasone exerted a better treatment effect which is attributed to the extraordinary ability of dexamethasone to decrease the IFP. In short, this work has demonstrated that the developed MSN-siRNA/CrPEI delivery system has great promise for the delivery of siRNA in tumor therapeutics.
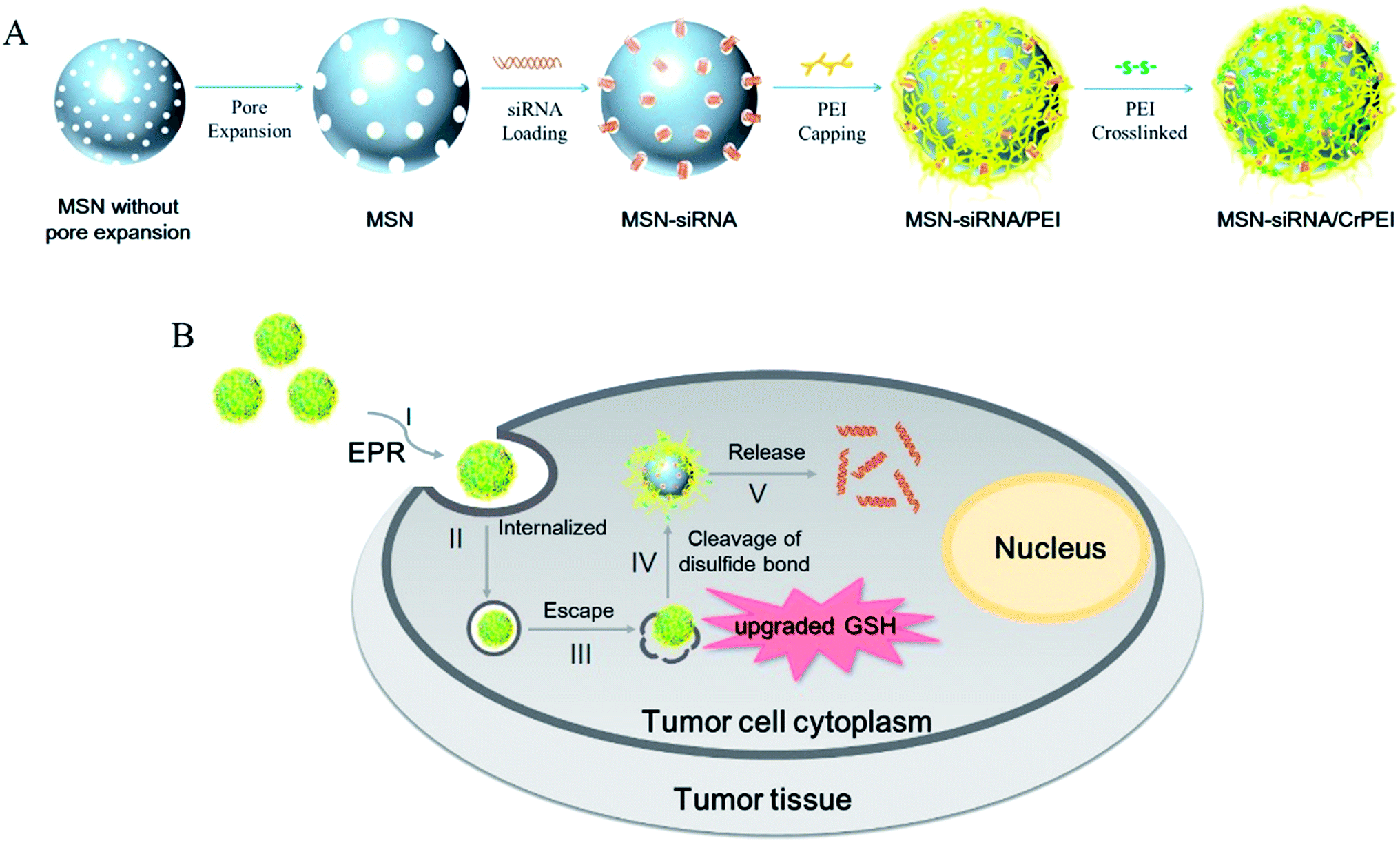 | ||
| Fig. 1 (A) Scheme illustrating the preparation of MSNs as delivery vectors loading siRNA. The process includes pore expansion, encapsulating siRNA molecules into the mesopores of MSNs (MSN-siRNA), PEI-capping of the MSN-siRNA composite (MSN-siRNA/PEI), and crosslinking of PEI via the disulfide bond of DSP (MSN-siRNA/CrPEI). (B) Flowchart of tumor-triggered targeting siRNA delivery: (I) accumulation of nanoparticles via the enhanced permeability and retention (EPR) effect in tumor tissue; (II) internalization of nanoparticles into tumor cells; (III) endosomal escape of delivery vehicles; (IV) glutathione-triggered PEI removal via cleavage of disulfide bonds; (V) release of siRNA into the cytoplasm from the mesopores, and development of RNAi. | ||
Materials
Hexadecyltrimethylammonium bromide (CTAB) was purchased from Biofunction Co. Ltd (Beijing, China). Tetraethylorthosilicate (TEOS) was purchased from ZKTZ Chemical Technology Co. Ltd (Beijing, China). 1,3,5-Trimethylbenzene (TMB) and guanidine hydrochloride were purchased from Sinopharm Chemical Reagent Co. Ltd (Beijing, China). Branched polyethylenimine (PEI, 2 kDa) was purchased from Sigma-Aldrich (St. Louis, USA). Lomant’s reagent (DSP) was obtained from Thermo Scientific (Waltham, MA, USA). Dithiothreitol (DTT) was purchased from Amresco (OH, USA). The scrambled siRNA (siNC) and the FAM-labeled negative siRNA (FAM-siRNA) (antisense strand, 5′-ACGUGACACGUUCGGAGAATT-3′), as well as siRNA targeting VEGF (siVEGF, antisense strand, 5′-GAUCUCAUCAGGGUACUCCdTdT-3′) were from Genepharma (Shanghai, China). The siRNA are double-stranded RNA oligos containing 21 nt. All primers were synthesized by AuGCT Biotechnology (Beijing, China). Dexamethasone sodium phosphate (Dex) was purchased from Energy-chemical (Shanghai, China). RPMI-1640 medium, penicillin–streptomycin, trypsin and Hoechst 33258 were obtained from Macgene Technology (Beijing, China). LysoTracker Red was purchased from Invitrogen (Carlsbad, CA, USA). The human VEGF ELISA kit was from Raybiotech (Atlanta, Georgia, USA). The Reverse Transcription System and GoTaq® qPCR Master Mix were from Promega (Madison, Wisconsin, USA). Pisum sativum agglutinin (PSA) was purchased from Vector labs (Burlingame, CA, USA).Methods
Preparation of MSNs with enlarged pores
The synthesized silica nanoparticles were prepared according to the literature26 with a little modification. 0.02 mmol CTAB was dissolved in 640 g methanol and 960 g water. 4.5 mL 1 M NaOH solution together with 4 mL TEOS were added to the solution with vigorous stirring for 8 h followed by the mixture being placed overnight to obtain a white precipitate, which was centrifuged and washed with ethanol and water three times each to remove any remaining surfactant. Then, the synthesized silica nanoparticles were dispersed in ethanol and sonicated for 30 min. 25 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture (v/v) of water and TMB was added subsequently to prepare the silica nanoparticles with enlarged pores. The mixture was placed in a pressure reactor with a thick wall and kept at 140 °C for 4 days.
:1 mixture (v/v) of water and TMB was added subsequently to prepare the silica nanoparticles with enlarged pores. The mixture was placed in a pressure reactor with a thick wall and kept at 140 °C for 4 days.
The obtained powder was washed with ethanol and water five times each, and refluxed in 200 mL acidic ethanolic solution (100 mL ethanol added with 1 mL concentrated hydrochloric acid) overnight. Afterwards, the white product was filtered out, washed with ethanol and then dried in a vacuum oven for 12 h.
Preparation of MSN-siRNA/CrPEI
The encapsulation of siRNA into the MSNs was carried out under dehydrated conditions.15 In brief, 0.3 mg synthesized MSNs was dispersed in 100 μL ethanol by probe supersonic, followed by adding 10 μL 12 M guanidine hydrochloride salt (Guan-HCl) and 20 μL water solution of siRNA. The mixture was dispersed by vortexing for 30 s and then was continuously shaken at 100 rpm at 25 °C for 1 h. The amount of adsorbed siRNA was determined from the difference in concentration of siRNA in the solutions before and after the adsorption process by a Nanodrop 2000 spectrophotometer (Thermo Scientific, USA).Then, the siRNA-loaded MSNs (MSN-siRNA) were dispersed in PEI ethanol solution to form aggregates capped with PEI (MSN-siRNA/PEI). After vortexing for 30 s, the mixture was incubated at 25 °C for 20 min, then centrifuged at 5000 rpm for 10 min to remove the free PEI in the supernatant. The determination of PEI concentration in the ethanol solution was performed using the standard curve plotted by measuring the absorption value at 220 nm through a TU-1900 spectrophotometer. The amount of capped PEI could be calculated from the difference in concentration of PEI in the solutions before and after the PEI-capping process.
To achieve PEI crosslinked by disulfide bonds, 0.01 M DSP in dimethylsulfoxide (DMSO) was added to the preformed aggregates dispersed in ethanol.27 The solutions were mixed by vigorous vortexing and incubated at 25 °C for 30 min. Then, dispersion of the obtained MSN-siRNA/CrPEI aggregates was promoted by incubation in acidic deionized water under sonication for several minutes. The aggregates were later treated with deionized water to obtain the MSN-siRNA/CrPEI complex.
Characterization of MSNs
Nitrogen adsorption–desorption isotherms were measured at 77 K with 100 mg MSNs, using an Accelerated Surface Area and Porosimetry System (ASAP 2020). Before the measurements, all samples were degassed at 300 °C for 12 h. Surface area calculations were carried out via the Brunauer–Emmett–Teller (BET) multimolecular layer adsorption model, whereas the average pore sizes were calculated from the desorption branch by using the Barrett–Joyner–Halenda (BJH) model.Transmission electron microscopy (TEM) was carried out on a JEM-2100F transmission electron microscope. Samples were prepared in ethanol, and a drop of each suspension was placed onto the copper grid. The grids were then dried at room temperature for several hours before TEM observation.
The zeta potentials were measured via a Malvern Zetasizer Nano ZS (Malvern, UK) in automatic measurement position and laser attenuation.
In vitro release experiments
The PEI capping function was investigated by the release profiles of siRNA from the obtained MSN-siRNA/CrPEI. The release profiles of siRNA with and without dithiothreitol (DTT) at pH 7.4 were carried out as follows. Two parallel vials containing MSN-siRNA/CrPEI (2.0 mg) had been dispersed in 2.0 mL phosphate buffer (PBS) with pH 7.4 at 37 °C. After 16 h, 2 mL 10 μM DTT solution was added to one of the MSN-siRNA/CrPEI vials. At particular time intervals, 10 μL sample from the dissolution medium was collected and the same volume of fresh solution was added. After centrifugation at 12000 rpm for 3 min, the supernatant of the samples was analyzed by Nanodrop 2000 (Thermo Scientific, USA). Each assay was repeated in triplicate. siRNA release from MSN-siRNA/PEI was also carried out as a comparison.
Cell culture
KB cells were grown in a RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Sijiqing, China), 100 IU mL−1 penicillin and 100 mg mL−1 streptomycin. All cells were maintained in a 37 °C humidified incubator with a 5% CO2 atmosphere.Cellular uptake
The cellular uptake of the nanoparticles loaded with FAM-siRNA was confirmed by fluorescence detection. KB cells were seeded in six-well plates at a density of 5 × 105 cells per well in 2 mL complete 1640 medium for 24 h. The cells were rinsed with PBS and incubated with MSN-siRNA/PEI and MSN-siRNA/CrPEI containing FAM-siRNA (50 nM) in a serum-free medium. After incubation for 4 h at 37 °C, the cells were rinsed with cold PBS, trypsinized and washed three times with cold PBS containing heparin (125 U mL−1). The samples were centrifuged, resuspended and determined immediately by flow cytometry.Intracellular trafficking and endosomal escape
A confocal fluorescence microscope was used to compare the intracellular distribution of the nanoparticles. KB cells were seeded on glass-bottom dishes at a density of 2.5 × 105 per well containing complete 1640 medium for 24 h. After three washes with PBS, the nanoparticles were treated in a serum-free medium for 4 h at 37 °C. The final concentration of FAM-siRNA in the culture medium was 50 nM. Subsequently, the cells were rinsed three times with cold PBS containing heparin (125 U mL−1) and fixed with 4% formaldehyde for 15 min at room temperature. Following another three rinses with cold PBS, the cell nuclei were stained with Hoechst 33258 (5 μg mL−1) for 20 min at 37 °C. Then, the cells were imaged using a confocal laser scanning microscope (CLSM, Leica, Heidelberg, Germany). FAM-siRNA and Hoechst 33258 were excited using 488 nm and 345 nm lasers, respectively.To track the internalization and endosomal escape of FAM-siRNA, KB cells were incubated for 0.5 h, 1 h, 2 h, 3 h, and 4 h with MSN-siRNA/CrPEI. Then, endosome/lysosome labeling was performed by LysoTracker Red (250 nM) for 30 min. After nuclear staining, the cells were observed by a CLSM. LysoTracker Red was excited by a 561 nm laser.
Cytotoxicity evaluation
The cytotoxicity of the nanoparticles (MSN/CrPEI) was evaluated via MTT assay. KB cells were seeded on 96-well plates at a density of 1 × 105 cells per well in 180 μL complete 1640 medium for 24 h. Then, cells were exposed to a fresh culture medium containing MSN/CrPEI of different concentrations ranging from 10 μg mL−1 to 100 μg mL−1 for an additional 48 h, followed by staining with MTT (5 mg mL−1). After 4 h incubation at 37 °C, the growth medium was replaced with 200 μL DMSO. Absorbance was measured at 570 nm through an iMark™ microplate absorbance reader (BioRad, USA).In vitro siRNA transfection and gene expression
In vitro RNAi experiments were carried out according to our previously work.19 For VEGF protein assessment, KB cells were seeded into six-well plates at a density of 2 × 105 per well. The amount of silencing VEGF in the supernatant medium was quantified by a human VEGF immunoassay kit (RayBiotech, USA) according to the instructions of the manufacturer. The total protein concentration was determined using a BCA Protein Assay Kit. For the cellular level of VEGF mRNA, a quantitative real-time polymerase chain reaction (qRT-PCR) method was applied. KB cells were seeded into 25 cm2 culture flasks at a density of 1 × 106 per well for 24 h. The final concentration of siRNA (VEGF siRNA or siNC) employed in the experiment was 80 nM. All experiments were performed in triplicate.In vivo tumor growth inhibition and body weight change
The anti-tumor efficacy was investigated using the xenograft tumor model. All animal procedures were performed with the approval of the Institutional Animal Care and Use Committee at Peking University Health Science Center. Female BALB/c nude mice (16 ± 2 g) were purchased from Vital River (Beijing, China), and kept under standard housing conditions with free access to standard food and water. KB cells (5.0 × 106) were implanted subcutaneously in the right armpit of the nude mice. When the tumor volume reached approximately 100 mm3, the mice were randomly divided into two groups (n = 5 per group) and treated with 50 μL 5% glucose (control) and MSN-siRNA/CrPEI via peritumoral injection, respectively. The dose of VEGF siRNA was 1.0 mg kg−1. Injection was performed every two days a total of three times. The anti-tumor effect was evaluated in terms of the tumor volume which was calculated by the formula: volume (mm3) = a × b2/2, where a and b are the major axis and minor axis of the tumor, respectively, as reported previously.28 After 6 days, the mice were sacrificed and the tumors were harvested, weighed and photographed. The body weight of each mouse was monitored every day.Interstitial fluid pressure
Interstitial fluid pressure (IFP) in the KB tumors was measured using an established wick-in-needle technique before tumor resection.29 Four nude mice per group were anesthetized by sodium pentobarbital solution and 2 to 3 measurements per tumor were made. The IFP was recorded and calculated as the pressure change from the baseline to the average maximum pressure measured after more than 10 seconds.30Perfusion and microvessel density observation
The perfusion analysis was carried out by referring to the literature.30 Three nude mice bearing tumors per group received an intravenous injection of fluorescence-Pisum sativum agglutinin (PSA). One hour later, the mice were euthanized. Tumor sections were frozen and cut into 4 μm thick sections. All frozen sections were analyzed using CLSM.To observe the intratumoral microvessel density, frozen sections of the harvested tumors were fixed in 4% paraformaldehyde. After being blocked with goat serum (10% in PBS), the sections were further treated with CD31 antibody (Abcam, UK) followed by a fluorescent secondary antibody and Hoechst 33258. All frozen sections stained with CD31 were analyzed using CLSM.
Determination of intratumoral VEGF content
To determine the intratumor VEGF expression, approximately 100 mg tumor tissue was treated for total protein and mRNA extraction followed by ELISA and qRT-PCR assay, respectively. Tumor tissue was homogenized and incubated with 600 μL RIPA tissue lysis buffer (1% 100 mM PMSF), and then centrifuged for 10 min at 12000 rpm. The total protein and VEGF protein concentration were determined by a BCA Protein Assay Kit and human VEGF immunoassay kit (RayBiotech, USA), respectively. To evaluate the levels of VEGF mRNA, each extracted mRNA sample was normalized to the same 260 nm absorbance value and detected via qRT-PCR as described above.
Dexamethasone administration
After the tumor volume reached approximately 100 mm3, dexamethasone (Dex) was administrated intravenously on days 1, 2, 3 and 5. Meanwhile, a siRNA-loaded formulation was administrated peritumorally on days 3, 5 and 7. The dose of siRNA and dexamethasone was 1.00 mg kg−1 and 3 mg kg−1, respectively. After 8 days, the mice were sacrificed and the tumors were harvested, weighed and photographed. The tumor volume, IFP, blood perfusion, microvessel density, intratumoral VEGF contents and the body weight were evaluated as described above.Statistical analysis
All data were analyzed using the student’s t-test in statistical evaluation. A P value < 0.05 was considered to indicate statistical significance (*P < 0.05, **P < 0.01).Results and discussion
Characterization of MSNs
MSNs with homogeneous size and morphology were prepared using TEOS as the silica source and cationic surfactant CTAB as the structure directing agent. The MSNs possessed enlarged pores after using TMB as a hydrophobic swelling agent. TMB has a suitable size making it possible for it to penetrate into the micelle rods inside the mesopores, driven by the hydrophobic forces between the surfactant alkyl chain and TMB, thus enlarging the hydrophobic core of the micelles and the pores while retaining the morphology and monodispersity of the mesopores.14 The synthesized MSNs had an average particle diameter of around 160 nm (Fig. 2A), BET surface area of 771.65 m2 g−1, BJH pore size of around 5.7 nm (Fig. 2B) and pore volume of 1.67 cm3 g−1 (Fig. 2C), much bigger than the traditional type of MCM-41 which has pore sizes of around 2.7 nm and pore volumes of 1 cm3 g−1.14 Fig. 2D shows the nitrogen sorption isotherm. At a relative pressure of 0.6 < P/P0 < 0.8, a steep increase occurred and a desorption hysteresis loop appeared, which is a type IV for H1 hysteresis loop classified by IUPAC, which indicates that the material has ordered mesopores.31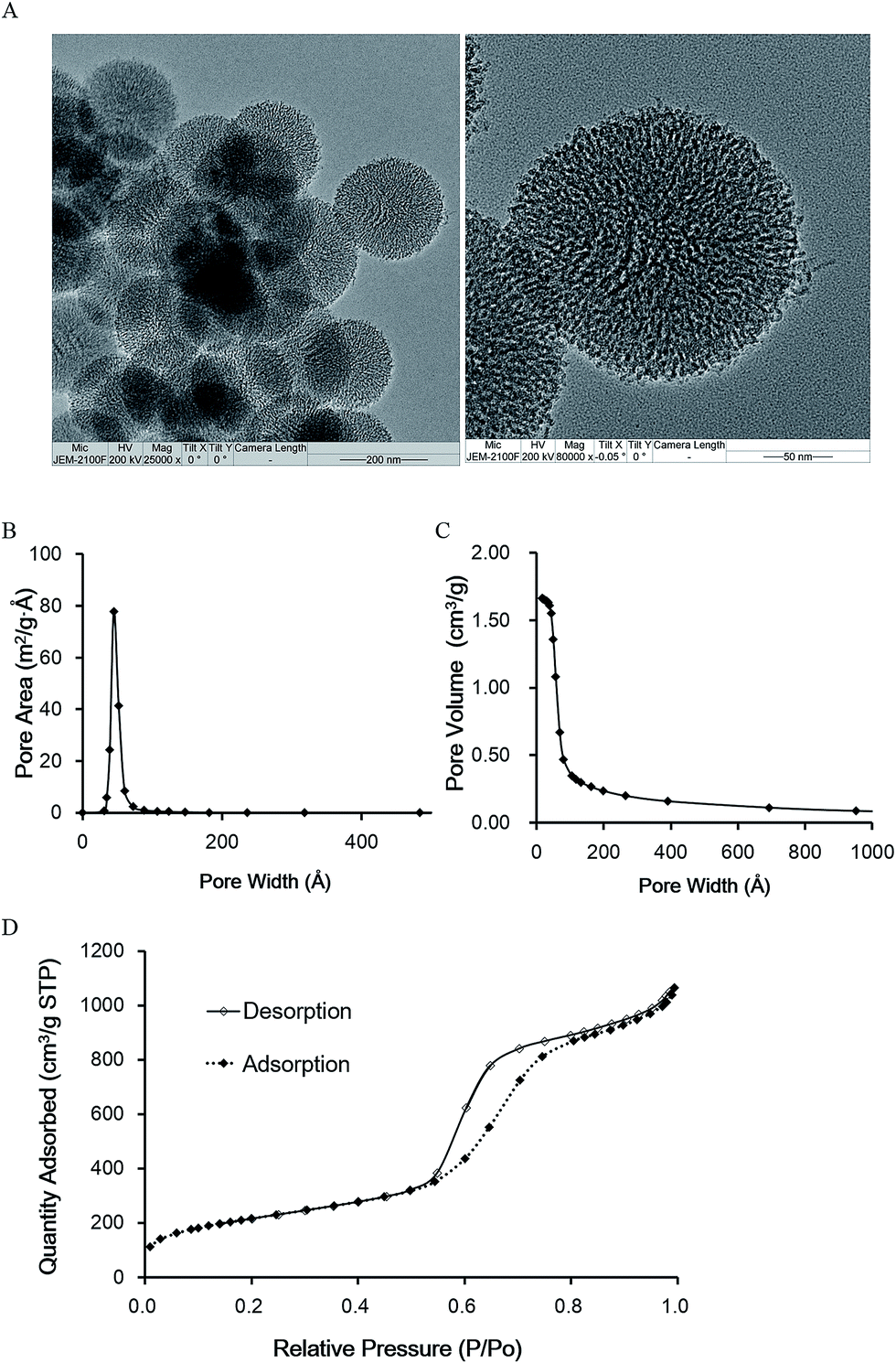 | ||
| Fig. 2 Characterization of MSNs. (A) TEM image of MSNs; (B) BJH desorption dA/dw pore area; (C) BJH desorption cumulative pore volume; (D) nitrogen adsorption–desorption isotherms. | ||
Characterization of MSN-siRNA/CrPEI
In hydrophilic solution, both siRNA and the MSN surfaces are negative and so are mutually repulsive. However, the previous literature reported that the high concentration of ions in the chaotropic salt solution could decrease the Debye length and shields the negative charges so as to weaken the repulsive electrostatic force between siRNA and the MSNs.15 Here, we utilized the published method with a little modification, using 0.923 M Guan-HCl and 76.9% ethanol (v/v) as the composition of the chaotropic salt solution. It is reported that Guan-HCl possesses the outstanding ability to catch water molecules, thus offering the shielded intermolecular electrostatic force, dehydration effect, and intermolecular hydrogen bonds to facilitate siRNA’s diffusion into the mesoporous structure, while organic solvents induce the dehydration effect to ensure that siRNA is encapsulated within the MSN mesopores.32 The loading efficiency of siRNA was about 65% and the amount of siRNA loaded by the MSNs reached 35 mg g−1 on average, which is higher than that of traditional loading of MSNs (27 mg siRNA per g MSNs).33 The higher loading capacity may result from the enlarged pores.To prepare PEI-coated MSNs, the siRNA-loaded MSNs were mixed with PEI in ethanol solution. Because siRNA was insoluble in ethanol, the PEI solution had to be sufficiently hydrophobic so that adsorbed siRNA would not desorb from the pores.15 The amount of PEI coated on the MSNs was 70 μg mg−1, which was calculated from the difference in PEI concentration in the solutions before and after the PEI-coating process via the standard curve of PEI concentration in ethanol.
The MSNs coated with PEI were subsequently crosslinked by the addition of the appropriate amount of DSP solution to introduce disulfide bonds by just a one step reaction to make the nanoparticles stable. The molar ratio of DSP and PEI was 2. Based on the calculated amount of PEI, the amount of DSP we needed was 8.10 × 10−8 mol g−1 MSNs.
Through every process the mesoporous silica was characterized by TEM. Fig. 3 shows the TEM micrographs of the vehicle during the different processes. Compared to the naked or bare MSNs (Fig. 3A), the mesoporous structures of MSN-siRNA (Fig. 3B), MSN-siRNA/PEI (Fig. 3C), and MSN-siRNA/CrPEI (Fig. 3D) gradually became fuzzy and the size of the nanoparticles changed from 175 nm (bare MSNs) to 210 nm (MSN-siRNA/CrPEI), which also confirmed that the MSNs had been successfully loaded with siRNA, embedded within the PEI layer and had a crosslinked PEI layer. The thickness of the PEI layer was about 15 nm.
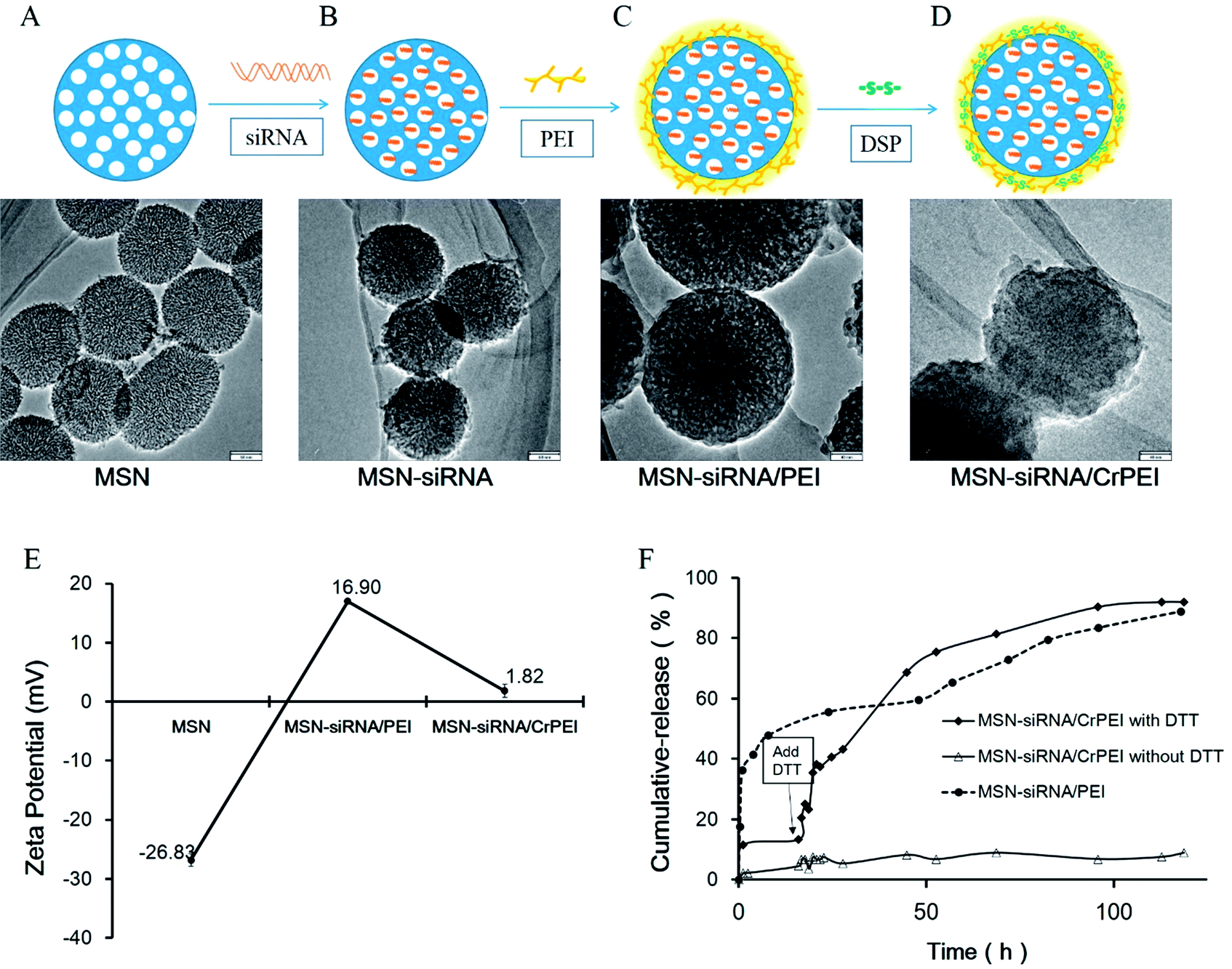 | ||
| Fig. 3 Characterization of MSN-siRNA/CrPEI. TEM images of (A) bare MSNs, (B) MSNs loaded with siRNA in the mesopores, (C) PEI capped MSN-siRNA, and (D) disulfide bond crosslinked PEI capped MSN-siRNA. The scale bar in A and B is 60 nm, and in C and D is 40 nm. (E) Zeta potential of various MSNs. (F) siRNA release profiles of MSN-siRNA/CrPEI and MSN-siRNA/PEI in phosphate buffer solution (PBS) at pH 7.4. | ||
The bare MSNs displayed a negative zeta potential (−(26.9 ± 1.05) mV, Fig. 3E). The positive zeta potential of MSN-siRNA/PEI (+(16.9 ± 0.30) mV) was provided by PEI because of its abundant primary amines. In addition, the zeta potential of MSN-siRNA/CrPEI (+(1.82 ± 1.08) mV) was much smaller than that of MSN-siRNA/PEI, indicating that the crosslinking of PEI via disulfide bonds resulted in many amide bonds that contributed to an acute reduction of the protonable primary amines of PEI, which may also decrease the toxicity of PEI on cells.
siRNA release
In order to evaluate whether the PEI crosslinked via disulfide linkages is sensitive to a reductive environment DTT, a small reductive molecule, was used to simulate the effects of GSH and was added to investigate siRNA release in vitro. As shown in Fig. 3F, no siRNA could be detected during 120 h in the absence of DTT, indicating the efficient confinement of siRNA in the mesopores of MSNs capped with PEI. In contrast, there was a sudden release of siRNA with the addition of DTT due to cleavage of the disulfide bonds. Under reductive conditions, the cumulative drug release percentage was about ∼90% at pH 7.4 after 120 h release, whereas for MSN-siRNA/PEI, the sudden release of siRNA was observed during the first hour, suggesting that the short chain PEI desorbed from the surface of the MSNs and released siRNA in an aquatic environment.In our study, we took advantage of short chain PEI (2 kD) that possesses less amino groups and positive charge than long chain PEI (25 kD), thus making desorption from the surface of the MSNs easier, which would cause siRNA release. The results suggest that the MSNs could be an effective siRNA carrier and encapsulate siRNA without release until the cap on the MSNs is removed by cleavage of the disulfide bonds induced by DTT or GSH in cells. This redox-triggered destabilization mechanism is the outcome of large differences in GSH concentration found in different tissues. In the circulation of blood, there is not enough reductant to cleave the disulfide bonds (the intracellular GSH level in normal tissue is 1–10 mM, but that in blood is 2 μM), while in tumor tissue environment GSH concentration is 7 to 10 times higher than that in normal cells.34 Thus, the nanoparticles were internalized into the tumor cells that offered a higher GSH level than the extracellular environment. As a consequence, it is much easier for the extremely elevated level of GSH to split the crosslinked PEI cap and release the cargo.
In brief, the prepared MSN-siRNA/CrPEI possesses high loading of siRNA and can be stimulated sensitively according to the changes in GSH in physiological compartments, protect siRNA from degradation in circulation and provide for timely drug release in tumor cells meaning they are “smart” multi-stage vesicles. In addition, the nature of MSN-siRNA/CrPEI is intended to bestow drug carriers with suitable properties for passive localization in tumor tissue by the EPR effect.35 All of these features suggested applications for drug delivery in specific biological compartments.
Cellular uptake, intracellular trafficking and endosomal escape
The poor membrane permeability, large MW (about 13.3 kDa) and hydrophilic nature of siRNA makes therapeutic application a difficulty.7 Two spectroscopy techniques were used to determine the cellular uptake, including flow cytometry and confocal fluorescence microscopy. Free FAM-siRNA and FAM-siRNA-loaded MSNs (MSN-siRNA/CrPEI and MSN-siRNA/PEI) in which the siRNA concentration was 50 nM, together with KB cells, were incubated for 4 hours at 37 °C. The results of flow cytometry are shown in Fig. 4A. Naked siRNA showed low florescence intensity, indicating little translocation of siRNA into cells, which was consistent with the nature of poor membrane permeability of naked siRNA. Both MSN-siRNA/CrPEI and MSN-siRNA/PEI presented significant uptake compared to that of the control and naked siRNA. Furthermore, MSN-siRNA/CrPEI showed a slight higher uptake of FAM-siRNA than that merely capped with PEI, suggesting more translocation of siRNA into the cells. This is perhaps due to electropositive PEI combining with electronegative cell membranes, making it easier for uncrosslinked short chain PEI to desorb from the MSN surface than the crosslinked PEI, resulting in greater release of siRNA during the entire process of internalization. The observations via confocal fluorescence microscopy as indicated in Fig. 4B were in accordance with those of flow cytometry. Some punctuate green fluorescence could be observed in the cytoplasm, suggesting that the nanoparticles had been internalized by cells.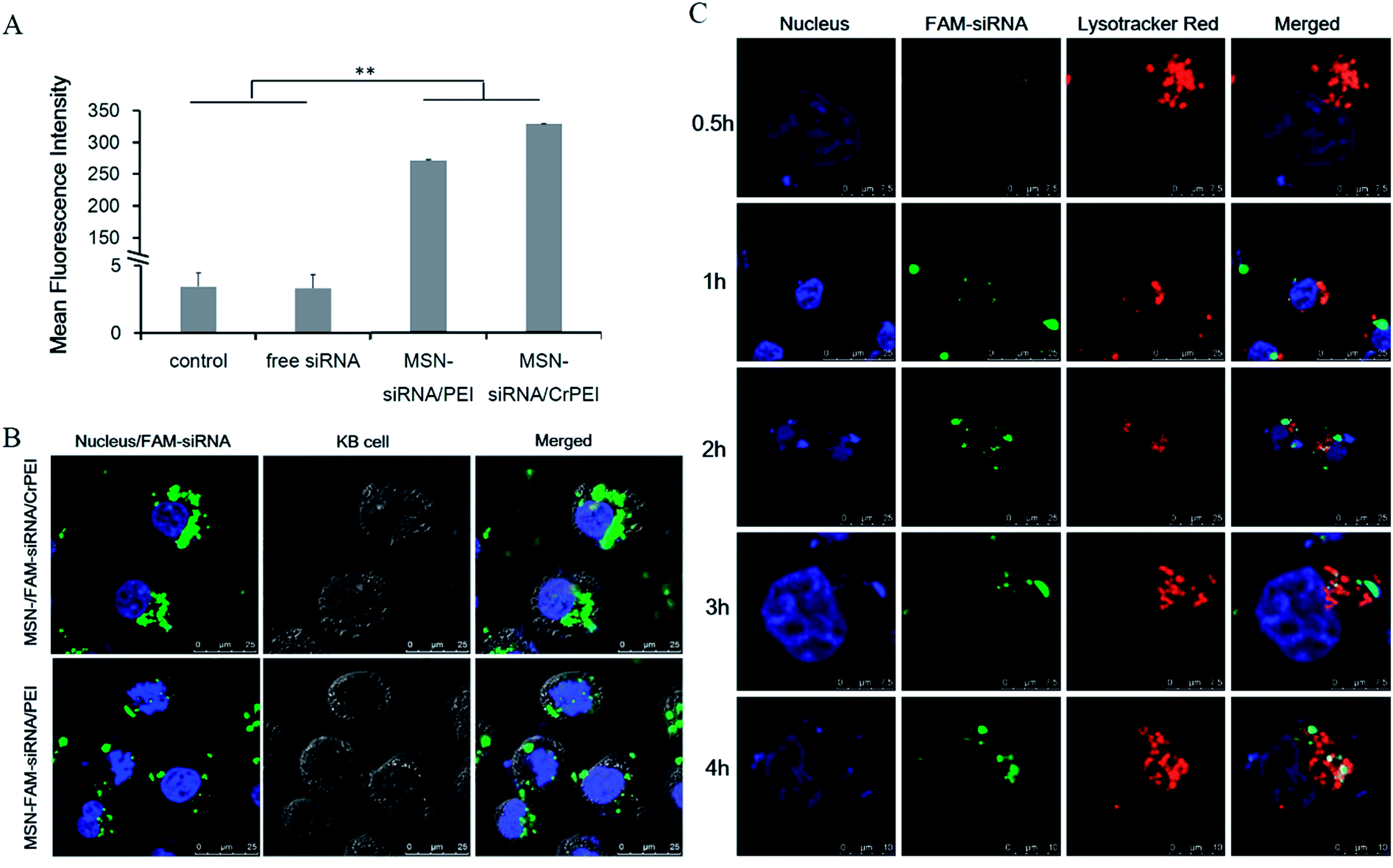 | ||
| Fig. 4 Cellular uptake and intracellular trafficking of FAM-siRNA delivered by MSNs. (A) Fluorescence intensity of KB cells incubated with free siRNA, MSN-siRNA/PEI and MSN-siRNA/CrPEI at 37 °C for 4 h as measured by flow cytometry. Cells treated with a serum-free media were used as the control. The data are expressed as the mean ± SD (n = 3). **P < 0.01. *P < 0.05. (B) Intracellular trafficking of FAM-siRNA (50 nM) delivered by MSNs incubated with KB cells for 4 h. (C) Intracellular distribution of MSN-siRNA/CrPEI in KB cells after being incubated for 0.5 h, 1 h, 2 h, 3 h and 4 h. Green, red and blue represent the fluorescence of FAM-siRNA, LysoTracker Red for endolysosomes and Hoechst 33258 for nuclei staining, respectively. | ||
In fact, to generate a substantial gene silencing effect, it is not only necessary for siRNA to be transferred into the cells, but also for escape from the endosome to work in the cellular cytoplasm. Thus, the intracellular distribution after trafficking into the cells and lysosome escape of siRNA was evaluated. KB cells were incubated with MSN-siRNA/CrPEI at 37 °C for different incubation times (0.5 h, 1 h, 2 h, 3 h and 4 h) followed by live cell imaging under a Leica SP5 confocal microscope. The endosome/lysosome was stained with LysoTracker Red. As shown in Fig. 4C, for the FAM-siRNA loaded MSNs capped with crosslinked PEI (MSN-siRNA/CrPEI), weak colocalization spots (yellow) of the green (FAM-siRNA) and red (LysoTracker Red) fluorescence could be found in the cytoplasm, indicating that the majority of encapsulated siRNA was within endolysosomes in the early phase of uptake (0.5 h). However, as time went on, the green fluorescence strengthened which suggested that the uptake increased and partially separated from red fluorescence over time (4 h), pointing to the successful escape of FAM-siRNA and distribution in the cytoplasm where siRNA mediated its function. This rapidly triggered escape involved the proton sponge mechanism of PEI.20
Effect of MSN-siRNA/CrPEI on cell activity
As we know, a critical issue for the application of any nanocarrier is the nanoparticles’ toxicity. Toxicity is associated with silanol groups on the surface of MSNs.11,36 The cytotoxicity of MSN/CrPEI and bare MSNs was evaluated through the MTT assay. Fig. 5 shows the viability of KB cells incubated with MSNs for 48 h. It was found that MSN/CrPEI exhibited little toxicity to cells, even though the dose of MSNs reached as high as 100 μg mL−1, while bare MSNs had toxic effects at high doses since slight inhibition of cellular viability appeared at a dose of 100 μg mL−1. The general consensus of the toxicity of MSN is related to the silanol groups that can hydrogen bond to membranes, or ionize to form SiO− which leads to strong interaction and possibly membranolysis. What is more, the silica surface produces radicals which react with water to yield reactive oxygen species (ROS) that also cause cell death.37 In addition, the toxicity was associated with the cell line in that the MSNs exerted different toxicities on different cell lines. In our other study, bare MSNs showed no significant toxicity on MCF-7 cells (data not shown). Fortunately the toxicity can be decreased by surface modification to protect the exposed silanol groups.38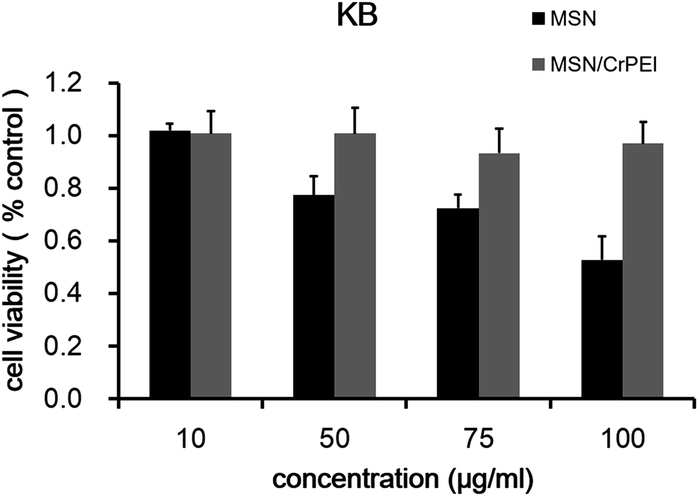 | ||
| Fig. 5 The survival rate of KB cells cultured with bare MSNs and MSN/CrPEI for 48 h. Data are presented as the mean ± SD (n = 5). | ||
It is known that PEI can induce interactions with cells and has toxicity, especially at higher doses.39 However, the previous study showed that crosslinked short chain PEI has a lower toxicity than the long chain21 due to the lower positive charge, which may explain why the MSNs capped with crosslinked PEI didn’t show much toxicity. The results showed that the nanocarriers were highly compatible with cell membranes under physiological conditions.
In vitro siRNA transfection and gene expression
We treated KB cells with loaded VEGF targeting siRNA MNSs and naked siRNA to verify gene suppression activity. As shown in Fig. 6A, compared with the control group, VEGF protein expression was significantly down-regulated in both MSN-siRNA/PEI (40%, P < 0.05) and MSN-siRNA/CrPEI (75%, P < 0.01), while the difference between the two preparation groups was thought to have led to a better protective efficiency of loading within MSN-siRNA/CrPEI than that of MSN-siRNA/PEI, resulting in a higher efficiency of siRNA delivery into cells and a better effect of target gene silencing.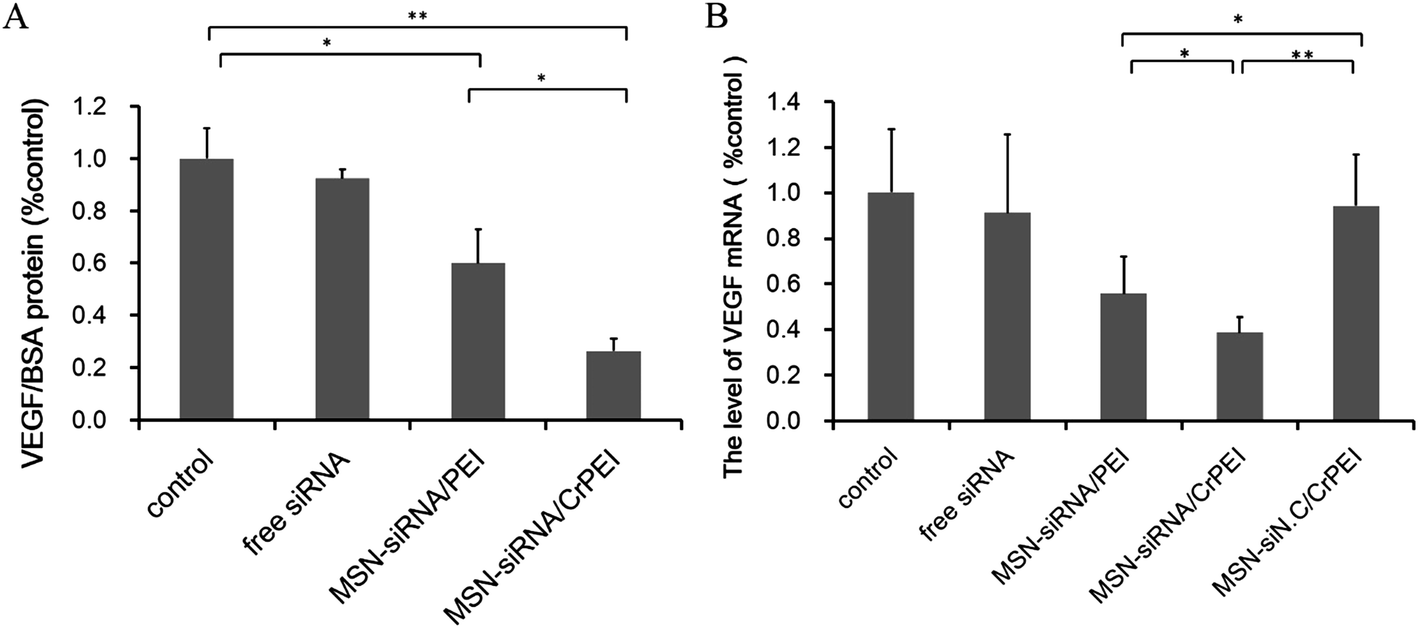 | ||
| Fig. 6 (A) The level of VEGF protein/BSA protein and (B) VEGF mRNA expression determined by ELISA and quantitative real-time PCR after culturing with various formulations carrying VEGF siRNA in KB cells, respectively. KB cells were transfected with various samples carrying VEGF siRNA or siNC at 37 °C for 4 h. Data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01. | ||
To confirm that the suppression of VEGF expression indeed resulted from VEGF RNA reduction mediated by VEGF siRNA in tumor cells, we adopted the method of qRT-PCR to detect the transcriptional VEGF mRNA level (Fig. 6B). The relative quantitative method of GAPDH, used as an internal reference, is applied to calculate the relative content of VEGF mRNA. The results were consistent with those of VEGF protein expression. Treatment with MSN-siRNA/CrPEI caused a greater down-regulation of VEGF mRNA (∼74%) compared with MSN-siRNA/PEI (∼40%), whereas there was no apparent knockdown shown by naked VEGF siRNA and MSN-siNC/CrPEI. The results were in accordance with the above assays of cellular uptake and endosomal escape, suggesting efficient delivery of bioactive siRNA into the cytosol.
In vivo tumor growth inhibition study
As expected, MSN-siRNA/CrPEI was shown to be effective in vitro. Therefore, the in vivo effects of nanoparticles loaded with siRNA was evaluated further to observe the therapeutic effect. Peritumoral administration of MSN-siRNA/CrPEI (1.00 mg kg−1 VEGF siRNA) was performed on nude mice bearing a KB xenografted tumor, and compared with the control. As shown in Fig. 7, MSN-siRNA/CrPEI showed a higher inhibition of tumor growth (Fig. 7A) and smaller tumor size (Fig. 7B) and weight of the excised tumors (Fig. 7C) compared with the control group.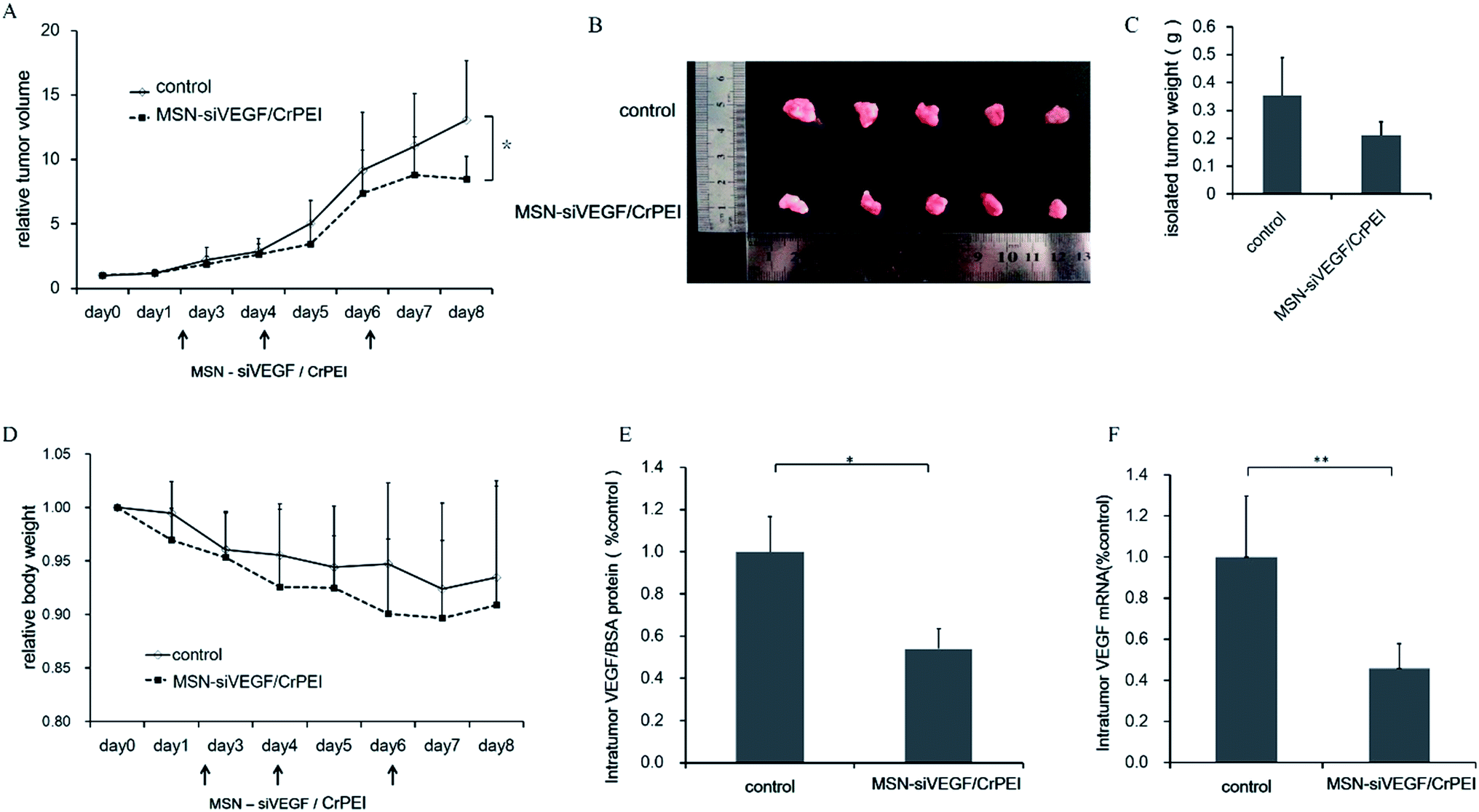 | ||
| Fig. 7 (A) In vivo antitumor study of MSNs in KB tumor-bearing BALB/c nude mice after injection with 5% glucose (control) or MSN-siVEGF/CrPEI. The dose of siRNA was 1.00 mg kg−1. Arrows represent the times of drug administration. Data are presented as the mean ± SD (n = 5–7). *P < 0.05 as compared with controls. (B) Photograph and (C) the weight of the solid tumors removed from different treatment groups at the study termination. Data are presented as the mean ± SD (n = 5). (D) The body weight variation of BALB/c nude mice implanted with KB cells after treatment. Data are presented as the mean ± SD (n = 5). There were no significant differences (P > 0.05). (E) The expression level of intratumor VEGF protein and (F) mRNA in tumors. Data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01. | ||
During the entire process of medication, the body weight of both the control group and treatment group showed a slight decline as shown in Fig. 7D, but there was no significant difference between the two groups. The reason for weight loss should be that the growth of the tumor took in a large part of nutrition and some factors secreted by tumor tissue would also cause an inflammatory response, thus having a negative impact on the mices’ living situation.
To evaluate whether the reduced tumor growth was associated with VEGF gene silencing in vivo, VEGF expression at both the protein and mRNA levels within the tumor was determined. As shown in Fig. 7E and F, the VEGF protein and mRNA levels of the MSN-sRNA/CrPEI group were significantly inhibited by about 46% (P < 0.05) and 55% (P < 0.01), respectively, compared with the control.
MSN-siVEGF/CrPEI reduces interstitial fluid pressure, induces angiogenesis regression, and increases vascular perfusion in tumors
Normal tissues have an IFP close to 0 mm Hg, whereas an elevated IFP is found in experimental and human tumors,40,41 which is contributed to by the absence of a normal lymphatic system, the abnormally high permeability within the tumor vessels and the increased propensity of microthrombus formation in both extravascular and vascular tumors. The higher osmotic pressure is not conducive to the accumulation of antineoplastic drugs within the tumor tissue, thus the modification of interstitial hypertension is considered an important factor in antineoplastic therapy. MSN-siRNA/CrPEI treatment reduced the IFP by 30% (Fig. 8A). The decrease indicated that the medication could mediate tumor vascular normalization which improves blood perfusion.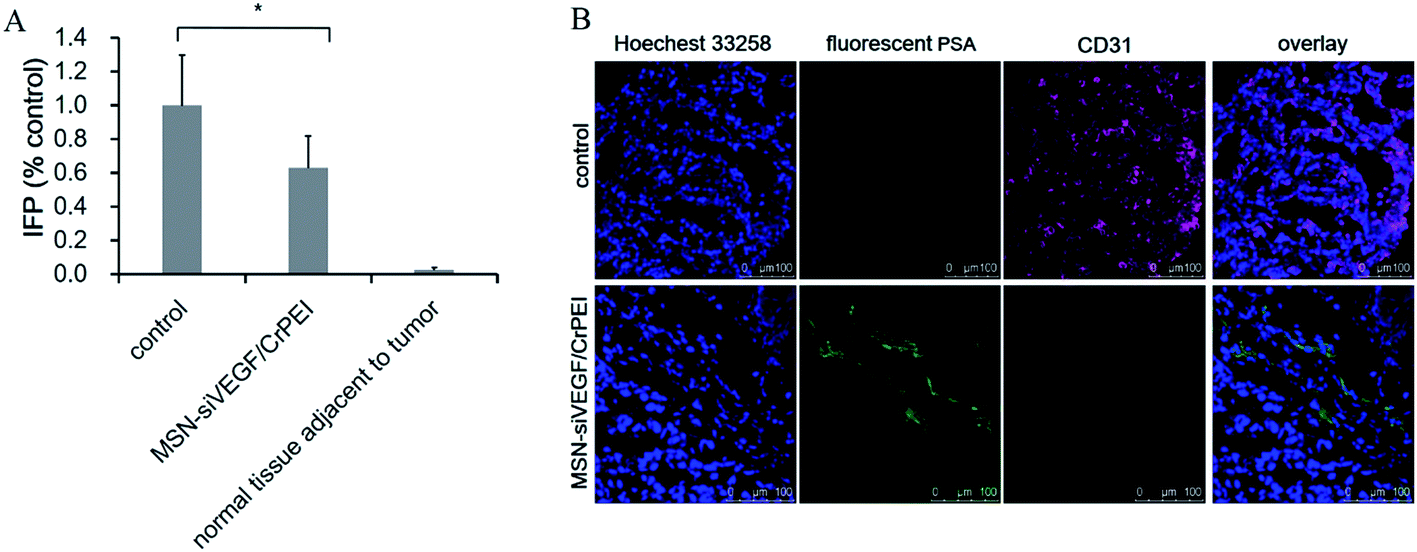 | ||
| Fig. 8 (A) IFP in KB tumor-bearing BALB/c nude mice after injection with 5% glucose (control) or MSN-siVEGF/CrPEI. The dose of siRNA was 1.00 mg kg−1. Data are presented as the mean ± SD (n = 4–5). *P < 0.05 as compared with the control. (B) The immunohistochemistry of CD31 and fluorescent PSA represented vessels and perfusion. | ||
Immunofluorescent staining of tumor sections for the analysis of tumor vasculature and blood perfusion was performed. As indicated in Fig. 8B, the control group had abundant CD31-positive tumor vessels and little blood flow. In contrast, a significant reduction of the CD31-positive tumor vessels and enhancement of blood flow marked by fluorescent PSA were observed in the VEGF siRNA treatment groups.
In tumor tissues, the physiological abnormalities associated with tumor vessels stimulate the overproduction of proangiogenic factors (i.e. VEGF).42,43 Proangiogenic factors promote the formation of new immature, tortuous, and hyperpermeable vessels,43 with disordered endothelial cells, loosely attached pericytes, and aberrant distribution of the vascular basement membrane.44,45 These abnormalities lead to an impaired blood flow.46 Precious literature47 elucidated that the observed effect of anti-VEGF therapy in preclinical models is a result of both vascular regression and normalization. Vascular regression is the growth reduction of immature vessels. The vascular normalization is characterized as vessel remodeling by the improvement of hyperpermeability, increased vascular pericyte coverage, a more ordered basement membrane, a reduction in tumor hypoxia and IFP and improved tumor vessel perfusion and oxygenation.45
All of the results revealed that VEGF siRNA in the MSNs had been delivered into the tumor and exerted effects, resulting in tumor vascular normalization, the increase of perfusion and the reduction of IFP, which were linked to a reduction in tumor growth (Fig. 7).
siRNA combined with dexamethasone administration for in vivo tumor growth inhibition
As we mentioned above, abnormalities associated with vasculature in tumors contribute to the high IFP, acting as an obstacle for the delivery of macromolecules into solid tumors.40 A lower IFP may lead to an improvement in the delivery and efficacy of exogenously administered therapeutics. Dexamethasone (Dex), a glucocorticoid class steroid hormone, is widely used as a potent anti-inflammatory and bone growth steroid or chemotherapeutic drug to treat childhood leukemia.48 Dex has been also reported for its strong capability to decrease the IFP of mice bearing tumors.40 So, Dex was given to KB tumor-bearing BALB/c nude mice to determine whether the combination of Dex and siRNA nanoparticles can improve the cancer therapy.As shown in Fig. 9A, the Dex group showed a reduced IFP by almost 80% compared to the control, indicating that Dex has strong capability to decrease the IFP. The combined administration groups (MSN-siRNA/CrPEI + Dex or MSN-siNC/CrPEI + Dex) both showed lower IFPs than the MSN-siRNA/CrPEI group, indicating a stronger IFP reduction.
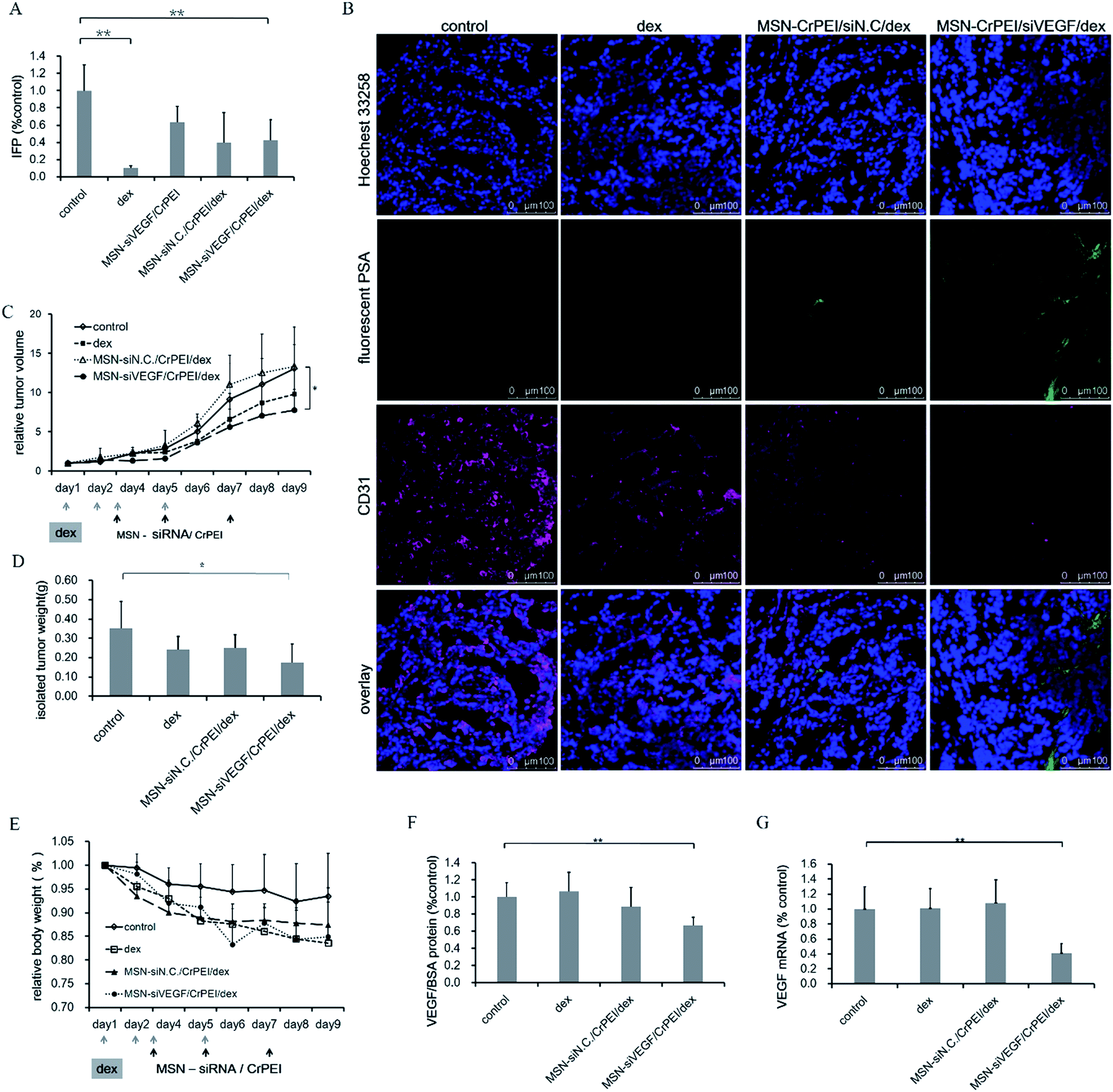 | ||
| Fig. 9 Effects of dexamethasone combination with MSNs in KB tumor-bearing BALB/c nude mice. The dose of siRNA and Dex was 1.00 mg kg−1 and 3 mg kg−1, respectively. (A) IFP. Data are presented as the mean ± SD (n = 4 or 5), *P < 0.05, **P < 0.01 as compared with the control. (B) Immunohistochemisty of CD31 (red) and fluorescent PSA (green) represented vessels and perfusion, respectively. (C) Relative tumor volumes. Arrows represent the time of drug administration. Data are presented as the mean ± SD (n = 5–7). *P < 0.05, **P < 0.01 as compared with the control. (D) Weight of the removed tumors. Data are presented as the mean ± SD (n = 5). *P < 0.05, **P < 0.01 as compared with the control. (E) Body weight variation of BALB/c nude mice implanted with KB cells after treatment. Data are presented as the mean ± SD (n = 5–7). There were no significant differences (P > 0.05). (F) The expression level of intratumor VEGF protein and (G) mRNA in tumors. Data are presented as the mean ± SD (n = 3). *P < 0.05, **P < 0.01 as compared with the control. | ||
The results of immunofluorescent staining (Fig. 9B) suggested that most tumor cells of the control group, Dex group and MSN-siNC/CrPEI with Dex group had a higher expression of CD31, accounting for a large number of new blood vessels that may distribute in the tumor tissue. The MSN-siRNA/CrPEI combined with Dex administration group had a lower vascular positive rate, showing that VEGF siRNA played a role in inhibiting the formation of tumor angiogenesis. Meanwhile, compared with the control group, the siRNA treatment group had relatively high blood perfusion marked with fluorescent-PSA and a tendency to normal, which is consistent with the determination results of decreased IFP.
As a consequence of the lower IFP, combined administration showed the highest inhibition of tumor growth and smallest tumor size and weight of excised tumors compared with other groups (Fig. 9C and D). The MSN-siVEGF/CrPEI group took second place. Although applying Dex alone also showed a trend of inhibition of the tumor growth, there was no significant difference compared to the control, indicating that siRNA is the dominant drug for solid tumor therapy. During the entire process of medication, the body weight of all groups declined (Fig. 9E), especially for the Dex group. According to the literature,40 a dosage of Dex ranging from 0–30 mg kg−1 should be safe, except the steroid toxicity, which may result in the phenomenon where the tumors are eaten by the host animals or their peers. However, during the process of the experiments, we found that a dosage of 3 mg kg−1 was high enough to induce the death of mice compared with the group that received only siRNA.
VEGF protein expression and mRNA levels within the tumors were determined to evaluate if combined administration could have a greater effect on VEGF gene silencing in vivo. As shown in Fig. 9F, the VEGF protein and mRNA levels of the combined administration group and MSN-siRNA/CrPEI group didn’t show much difference. The results also suggested that siRNA should be the initiator for adjusting mRNA and protein expression and for mediating the gene silencing.
Although the antitumor effects of the combined administration of Dex and MSN-siRNA/CrPEI are the best, they are still a long way from the ideal effect. A decreased IFP and improved perfusion may contribute to the EPR effect,44 which provides a greater opportunity for more nanoparticles to accumulate in the tumor. However, it is undeniable that long-term systemic exposure to Dex causes adverse side effects including slowed growth, stomach and intestinal bleeding caused by ulcers, damage to the joints, high blood sugar (Cushing’s syndrome), high blood pressure (hypertension) and of the most importance, immunosuppression, due to nonspecific killing of normal T and B lymphocytes.48 So, it is necessary to further study the treatment effects of Dex and balance the benefits and risks.
VEGF-siRNA showed moderate anti-tumor efficacy and single VEGF inhibition could not completely suppress angiogenesis and tumor growth. Furthermore, the effect of siRNA is temporary.49 However, it is of the most importance that antiangiogenic, specifically anti-VEGF, therapy in tumors can normalize vasculature that can provide a window of opportunity for concurrent chemotherapy, which acts as the main effect of cytotoxicity, and collaborative work to generate the highest anti-tumor efficacy via multiple tumor inhibition pathways.
Conclusion
In summary, we have successfully prepared MSNs capped with disulfide bond crosslinked PEI as nanoparticles to deliver siRNA. MSN-siRNA/CrPEI possessed considerable siRNA loading capability, ascendant sensitivity to the reductive environment, negligible cytotoxicity to cells, ready internalization into cells and efficient escape from endolysosomes, so that the nanoparticles release the loaded siRNA molecules in the cytoplasm and mediate significant target gene silencing effects in tumor cells. As a result, the developed MSN-siRNA/CrPEI delivery system was able to facilitate anti-tumor efficacy, gene silencing and IFP reduction in KB xenografted tumors based on anti-angiogenesis and vascular normalization effects in vivo by targeting VEGF. In addition, the combined administration of the MSN-siRNA/CrPEI delivery system with dexamethasone exerted a better treatment effect which is attributed to the strong ability of dexamethasone to decrease the IFP, and a lower IFP leads to an improvement in the delivery and efficacy of exogenously administered therapeutics. It is believed that the present MSN-siRNA/CrPEI delivery system can be further developed as an effective strategy for siRNA delivery.Acknowledgements
We would like to acknowledge the National Key Basic Research Program (no. 2013CB932501), NSFC (no. 81273454, 81473156), Beijing NSF (no. 7132113) and Doctoral Foundation of the Ministry of Education (no. 20130001110055) for funding of the work.References
- D. Hanahan and J. Folkman, Cell, 1996, 86, 353–364 CrossRef CAS.
- H. M. Verheul, H. Hammers, K. van Erp, Y. Wei, T. Sanni, B. Salumbides, D. Z. Qian, G. D. Yancopoulos and R. Pili, Clin. Cancer Res., 2007, 13, 4201–4208 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- C. G. Willett, Y. Boucher, E. di Tomaso, D. G. Duda, L. L. Munn, R. T. Tong, D. C. Chung, D. V. Sahani, S. P. Kalva, S. V. Kozin, M. Mino, K. S. Cohen, D. T. Scadden, A. C. Hartford, A. J. Fischman, J. W. Clark, D. P. Ryan, A. X. Zhu, L. S. Blaszkowsky, H. X. Chen, P. C. Shellito, G. Y. Lauwers and R. K. Jain, Nat. Med., 2004, 10, 145–147 CrossRef CAS PubMed.
- A. R. Kirtane, S. M. Kalscheuer and J. Panyam, Adv. Drug Delivery Rev., 2013, 65, 1731–1747 CrossRef CAS PubMed.
- N. S. Vasudev and A. R. Reynolds, Angiogenesis, 2014, 17, 471–494 CrossRef CAS PubMed.
- L. Aagaard and J. J. Rossi, Adv. Drug Delivery Rev., 2007, 59, 75–86 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2010, 8, 129–138 CrossRef PubMed.
- I. I. Slowing, J. L. Vivero-Escoto, C. W. Wu and V. S. Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- B. Liu, C. Li, P. Ma, Y. Chen, Y. Zhang, Z. Hou, S. Huang and J. Lin, Nanoscale, 2015, 7, 1839–1848 RSC.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- P. Selvam, S. K. Bhatia and C. G. Sonwane, Ind. Eng. Chem. Res., 2001, 40, 3237–3261 CrossRef CAS.
- H. Wen, J. Guo, B. Chang and W. Yang, Eur. J. Pharm. Biopharm., 2013, 84, 91–98 CrossRef CAS PubMed.
- M. Mizutani, Y. Yamada, T. Nakamura and K. Yano, Chem. Mater., 2008, 20, 4777–4782 CrossRef CAS.
- X. Li, Q. R. Xie, J. Zhang, W. Xia and H. Gu, Biomaterials, 2011, 32, 9546–9556 CrossRef CAS PubMed.
- D. V. Svintradze and G. M. Mrevlishvili, Int. J. Biol. Macromol., 2005, 37, 283–286 CrossRef CAS PubMed.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- P. Horcajada, A. Rámila, J. Pérez-Pariente and M. Vallet-Regı, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS PubMed.
- Z. Z. Yang, J. Q. Li, Z. Z. Wang, D. W. Dong and X. R. Qi, Biomaterials, 2014, 35, 5226–5239 CrossRef CAS PubMed.
- A. Akinc, M. Thomas, A. M. Klibanov and R. Langer, J. Gene Med., 2005, 7, 657–663 CrossRef CAS PubMed.
- R. Deng, Y. Yue, F. Jin, Y. Chen, H. F. Kung, M. C. M. Lin and C. Wu, J. Controlled Release, 2009, 140, 40–46 CrossRef CAS PubMed.
- E. Fleige, M. A. Quadir and R. Haag, Adv. Drug Delivery Rev., 2012, 64, 866–884 CrossRef CAS PubMed.
- G. Saito, J. A. Swanson and K. D. Lee, Adv. Drug Delivery Rev., 2003, 55, 199–215 CrossRef CAS.
- G. Wu, Y. Z. Fang, S. Yang, J. R. Lupton and N. D. Turner, J. Nutr., 2004, 134, 489–492 CAS.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- M. H. Kim, H. K. Na, Y. K. Kim, S. R. Ryoo, H. S. Cho, K. E. Lee, H. Jeon, R. Ryoo and D. H. Min, ACS Nano, 2011, 5, 3568–3576 CrossRef CAS PubMed.
- M. Neu, O. Germershaus, S. Mao, K. H. Voigt, M. Behe and T. Kissel, J. Controlled Release, 2007, 118, 370–380 CrossRef CAS PubMed.
- W. Gao, B. Xiang, T. T. Meng, F. Liu and X. R. Qi, Biomaterials, 2013, 34, 4137–4149 CrossRef CAS PubMed.
- M. Kato, Y. Hattori, M. Kubo and Y. Maitani, Int. J. Pharm., 2012, 423, 428–434 CrossRef CAS PubMed.
- M. Murakami, M. J. Ernsting, E. Undzys, N. Holwell, W. D. Foltz and S. D. Li, Cancer Res., 2013, 73, 4862–4871 CrossRef CAS PubMed.
- X. Li, J. Zhang and H. Gu, Langmuir, 2011, 27, 6099–6106 CrossRef CAS PubMed.
- P. Chomczynski and N. Sacchi, Nat. Protoc., 2006, 1, 581–585 CrossRef CAS PubMed.
- X. Li, Y. Chen, M. Wang, Y. Ma, W. Xia and H. Gu, Biomaterials, 2013, 34, 1391–1401 CrossRef CAS PubMed.
- S. Aluri, S. M. Janib and J. A. Mackay, Adv. Drug Delivery Rev., 2009, 61, 940–952 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopoulos, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- I. I. Slowing, C. W. Wu, J. L. Vivero-Escoto and V. S. Y. Lin, Small, 2009, 5, 57–62 CrossRef CAS PubMed.
- M. A. A. Schoonen, C. A. Cohn, E. Roemer, R. Laffers, S. R. Simon and T. O’Riordan, Rev. Mineral. Geochem., 2006, 64, 179–221 CrossRef CAS.
- D. Tarn, C. E. Ashley, M. Xue, E. C. Carnes, J. I. Zink and C. J. Brinker, Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- J. Hoon Jeong, L. V. Christensen, J. W. Yockman, Z. Zhong, J. F. Engbersen, W. Jong Kim, J. Feijen and S. Wan Kim, Biomaterials, 2007, 28, 1912–1917 CrossRef PubMed.
- P. E. Kristjansen, Y. Boucher and R. K. Jain, Cancer Res., 1993, 53, 4764–4766 CAS.
- Y. Boucher and R. K. Jain, Cancer Res., 1992, 52, 5110–5114 CAS.
- M. Datta, L. E. Via, W. S. Kamoun, C. Liu, W. Chen, G. Seano, D. M. Weiner, D. Schimel, K. England, J. D. Martin, X. Gao, L. Xu, C. E. Barry and R. K. Jain, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1827–1832 CrossRef CAS PubMed.
- P. Carmeliet and R. K. Jain, Nat. Rev. Drug Discovery, 2011, 10, 417–427 CrossRef CAS PubMed.
- N. Bertrand, J. Wu, X. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed.
- P. Baluk, H. Hashizume and D. M. McDonald, Curr. Opin. Genet. Dev., 2005, 15, 102–111 CrossRef CAS PubMed.
- R. K. Jain, J. Clin. Oncol., 2013, 31, 2205–2218 CrossRef CAS PubMed.
- S. Goel, D. G. Duda, L. Xu, L. L. Munn, Y. Boucher, D. Fukumura and R. K. Jain, Physiol. Rev., 2011, 91, 1071–1121 CrossRef CAS PubMed.
- V. Krishnan, X. Xu, S. P. Barwe, X. Yang, K. Czymmek, S. A. Waldman, R. W. Mason, X. Jia and A. K. Rajasekaran, Mol. Pharm., 2012, 10, 2199–2210 CrossRef PubMed.
- Z. X. Lu, L. T. Liu and X. R. Qi, Int. J. Nanomed., 2011, 6, 1661–1673 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |