
BiOBr photocatalyzed decarboxylation of glutamic acid: reaction rates, intermediates and mechanism†
Yanfen Fangab,
Hongwei Yangab,
Wei Zhouab,
Yue Lic,
David M. Johnsonab and
Yingping Huang*ab
aInnovation Center for Geo-Hazards and Eco-Environment in Three Gorges Area, Yichang 443002, Hubei province, China. E-mail: huangyp@ctgu.edu.cn; Fax: +86 717 6395966; Tel: +86 717 6397488
bEngineering Research Center of Eco-environment in Three Gorges Reservoir Region, Ministry of Education, China Three Gorges University, Yichang, 443002, China
cDepartment of Chemistry, Nankai University, Tianjin 300071, P. R. China
First published on 19th June 2015
Abstract
The degradation of glutamic acid by BiOBr under both UV and visible irradiation was investigated and compared with degradation by TiO2/UV. Analysis of the reaction rates and the distribution of intermediates was used to show that both BiOBr systems, unlike the TiO2 system, catalyze direct substrate oxidation by valance band holes.
Visible light photocatalysis has attracted continuing attention because of potential applications in water purification.1,2 A recently developed photocatalyst, BiOBr, is an efficient visible light photocatalyst3–5 with visible light activity higher than that of N-doped TiO2.6 As described previously, BiOBr has two discreet valance bands produced from O-2p and Br-4p orbitals.7,8 The two bands respond, respectively, to UV and visible light excitation and holes of different oxidation potential are generated, providing multiple mechanisms for photocatalytic degradation.7
Amino acids are biologically important organic compounds, both as building blocks for proteins and as metabolic intermediates. As biodecomposition products, amino acids are distributed widely in natural waters.9,10 The concentration of amino acids in surface water is generally in the range of 2.5–60 nM.11,12 Although amino acids are nontoxic, they can form carcinogenic and mutagenic species during the water purification process.13,14 For example, amino acids were converted primarily to halomethanes and haloacetic acids by chlorination.15–17 More importantly, with naturally occurring toxins it is usually the carboxyl group of an amino acid that binds to the affected enzyme.18–21 For example, with the well-known cyanotoxin, microcystin-LR, the free carboxyl groups on D-Glu and D-MeAsp bind with the metal atom and Arg96 of protein phosphatase 1 (PP1) to inhibit protein phosphorylation.19,20 Thus, understanding the degradation mechanism of amino acids, particularly the decarboxylation process, is of practical significance for water purification.
Glutamic acid (Glu) is one of the proteinogenic amino acids and, with a second carboxyl group on the side chain, it is an ideal substrate for comparing the degradation process of carboxylic acids with that of amino acids. In this work, we used BiOBr as the photocatalyst to degrade Glu under both UV and Vis irradiation. The degradation process was examined with 1H NMR and 18O isotope labeling and spin trapping ESR were used to elucidate the reaction mechanism. These results were compared with those from a TiO2 system to show the effect of the valance band structure of BiOBr on the catalytic degradation of amino acids.
D2O suspensions containing BiOBr and Glu were irradiated with UV or visible light for a given time and then analyzed using 1H NMR analysis after removing the photocatalyst. Compared with parent substrate (Glu), the reacted solutions gave additional peaks at δ of 1.93, 2.48, 3.26 and 8.35 with both UV and visible light irradiated systems (Fig. 1). Using reference compounds, these peaks were assigned to acetic acid (AA), succinic acid (SA), malonic acid (MA) and formic acid (FA). The change in the 1H NMR spectrum with reaction time also provides the kinetics of substrate consumption and intermediate formation in the BiOBr/UV and BiOBr/Vis systems (Fig. 2).
 | ||
| Fig. 1 1H NHR spectra of oxidative products of Glu in BiOBr/Vis and BiOBr/UV systems, 1 g L−1 BiOBr, c0Glu = 10 mmol L−1, 10 mL D2O. | ||
 | ||
| Fig. 2 Concentration change of substrate and oxidation products during photocatalytic oxidation of Glu in (a) BiOBr/Vis system and (b) BiOBr/UV system. | ||
During photocatalytic oxidation, Glu forms SA initially and further reaction of the primary intermediate gives MA, AA and FA. However, no signal for aspartic acid, the decarboxylation product of Glu, was recorded, a clear indication that degradation begins with the amino group rather than the carboxyl. The different decarboxylation of Glu in BiOBr/Vis and BiOBr/UV systems was proposed in Scheme 1 (additional details are shown in Fig. S1 (ESI†)). Similarly, during the BiOBr photocatalyzed oxidation of microcystin-LR,22 degradation also begins with oxidation of the amino-carboxyl structure of Glu. These results indicate that the amino-carboxyl structure is susceptible to oxidation in BiOBr photocatalytic systems. Due to the reactivity of this structure, amino acids are more readily degraded by BiOBr than are free carboxylic acids.
 | ||
| Scheme 1 Difference in the decarboxylation of Glu in BiOBr/Vis (gray arrows) and BiOBr/UV (black arrows) systems. Solid arrows shows a major reaction route, dashed arrows represent a minor reaction route. | ||
The BiOBr/Vis and BiOBr/UV systems display different degradation kinetics, but the visible light irradiated system also shows lower selectivity for SA and a markedly higher selectivity for MA (Table 1). This phenomenon is attributed to the discrete valance band structure of BiOBr. The holes generated by UV (hO-2p+) and visible light (hBr-4p+) excitation have different oxidation potentials, leading to different secondary reactions and the observed differences in rate. The results obtained in these systems were also compared with those of the classic TiO2/UV system to show the unique properties of BiOBr photocatalysis. It was observed that the TiO2/UV photocatalyzed oxidation of Glu gave remarkably low intermediate concentrations. The total selectivity of SA and MA in the TiO2 photocatalyzed system is only 2.8%, which is much lower than that of BiOBr/Vis and BiOBr/UV systems (37.4% and 21.8%, respectively, Table 1). TiO2 has a valance band (Evb = 2.7 V) more oxidizing than either of the two valance bands of BiOBr and its hole oxidizes H2O to ˙OH. It was reported that ˙OH plays a significant role in TiO2 photocatalyzed degradation of amino acids.23–25 Considering the valance band potentials and differences observed between the BiOBr and TiO2 systems, we assume that the valance band hole of both BiOBr systems initiates the degradation of Glu by direct oxidation rather than by ˙OH mediated reactions.
| System | rda (mmol L−1 h−1) | rfb (mmol L−1 h−1) | Sel.c (%) | ||
|---|---|---|---|---|---|
| SA | MA | SA | MA | ||
| a Decomposition rate of Glu.b Formation rate of intermediate.c Ratio of consumption rate of substrate to accumulation rate of intermediate. | |||||
| BiOBr/Vis | 0.199 | 0.019 | 0.055 | 9.6 | 27.8 |
| BiOBr/UV | 1.071 | 0.152 | 0.081 | 14.2 | 7.6 |
| TiO2/UV | 13.33 | 0.197 | 0.173 | 1.5 | 1.3 |
Since the photocatalytic degradation of Glu starts from the amino-carboxyl end and leads initially to SA, the decarboxylated and deaminated product, we anticipated that the source of oxygen atoms in the carboxyl group formed in this process could give useful information about the mechanism of the reaction. These experiments were carried out in 18O-enriched water (H218O) and atmospheric 16O2. Samples from the three systems were collected at times that resulted in similar substrate conversion (20–30%), and analyzed by derivative GC-MS (Fig. S2–S4 (ESI†)). As shown in Table 2, O atoms from both H2O and O2 were incorporated into SA under BiOBr photocatalysis condition. The SA formed in BiOBr/UV and BiOBr/Vis systems have similar isotope abundances of carboxyl O atoms (16O% = 13–14), which illustrates that these two systems react with similar mechanisms. In contrast, the SA formed in the TiO2/UV system gave an 16O abundance (16O% = 6.4) less than half that of the BiOBr systems. TiO2 photocatalysis clearly incorporates more H2O derived oxygen to the product than the BiOBr systems. We also performed 18O2 isotope labeling experiments and similar results were obtained (Table S1 and Fig. S5–S7 (ESI†)). Since the valance band hole of TiO2 can oxidize H2O to ˙OH and incorporate O atoms from H2O to the product, the higher proportion of H2O derived oxygen in the TiO2/UV system is reasonable. These results also corroborate the direct oxidation mechanism proposed for BiOBr systems. We propose that, in both the BiOBr/UV and BiOBr/Vis systems, the photogenerated hole (hO-2p+ or hBr-4p+) oxidizes Glu to a cation radical, which then reacts with either O2 or H2O to produce the carboxyl group on the product.
| System | Time (min) | Substrate conv. (%) | SA yield (%) | Abundanceb (%) | |
|---|---|---|---|---|---|
| 16O2 | H218O | ||||
| a 1 g L−1 photocatalyst, c0Glu = 10 mmol L−1, 2 mL H218O.b Average value of the two O atoms of the formed carboxyl group, corrected with the oxygen isotope abundance of solvent H218O and the natural isotope abundance of aerial O2. | |||||
| BiOBr/Vis | 480 | 29.9 | 11.9 | 14.2 | 85.8 |
| BiOBr/UV | 90 | 23.8 | 23.8 | 13.1 | 86.9 |
| TiO2/UV | 20 | 20.8 | 20.8 | 6.4 | 93.6 |
To further confirm that direct oxidation of Glu accounts for the larger pool of intermediates and higher proportion of O2-derived oxygen in SA observed in the BiOBr systems, spin-trapping ESR spectroscopy was used to detect the formation of ˙OH. The results were again compared with those of TiO2 and are shown in Fig. 3. In contrast to the TiO2/UV system, the signals from trapped ˙OH recorded in the BiOBr systems was either weaker or nonexistent. Because neither of the valance band holes of BiOBr can oxidize H2O, the small amount of ˙OH is attributed to the reduction of O2 by conduction band electrons (O2 → ˙OOH → H2O2 → ˙OH) and ˙OOH was detected (Fig. S8 (ESI†)).
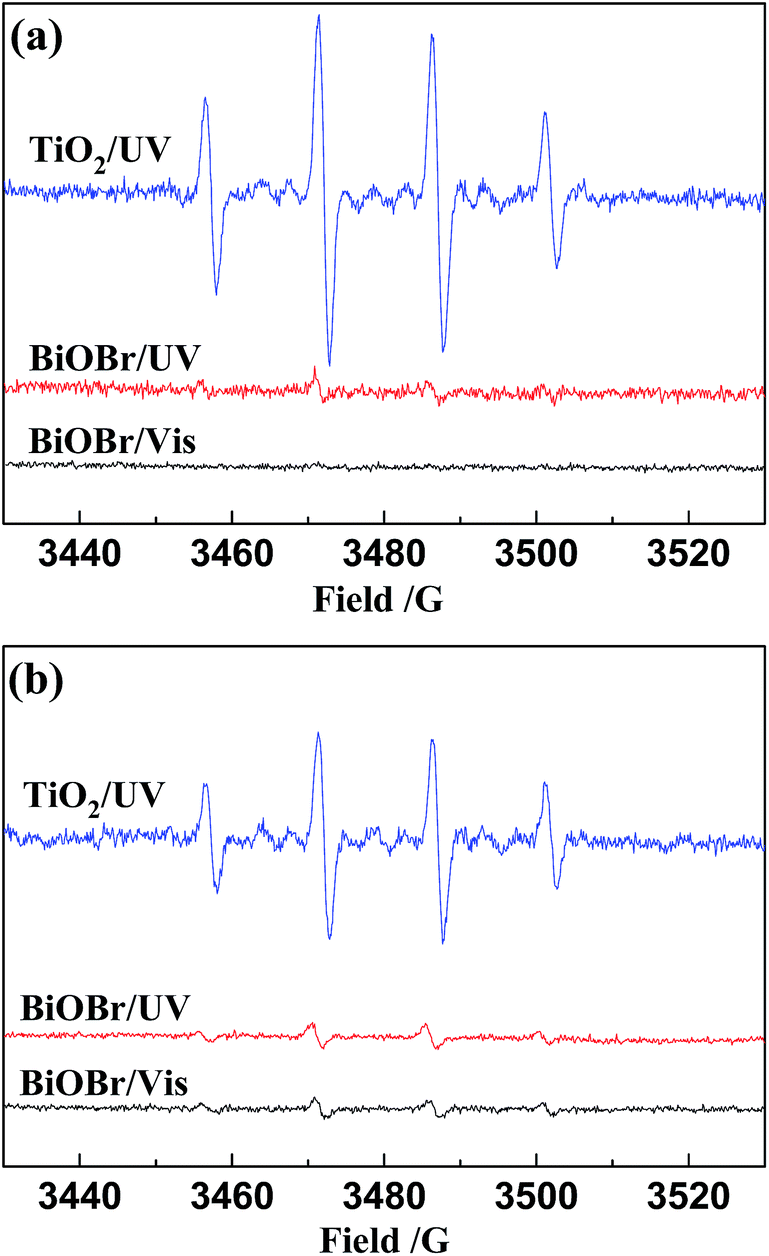 | ||
| Fig. 3 ESR signals of the DMPO-˙OH adducts in TiO2/UV, BiOBr/UV and BiOBr/Vis systems (a) without and (b) with Glu (10 mmol L−1). 1 g L−1 photocatalyst, cDMPO = 0.4 mol L−1. | ||
Conclusions
In summary, we studied the BiOBr catalyzed degradation of Glu under UV and visible light irradiation. Results indicate that, in both BiOBr/UV and BiOBr/Vis systems, the degradation process is initiated by direct substrate oxidation by the valance band hole. This, in turn, leads to the same primary product with the same source of oxygen in the carboxyl group formed on SA. However, the difference in the hole oxidation potentials of BiOBr/UV and BiOBr/Vis leads to different degradation rates, different secondary degradation processes and different distributions of degradation intermediates.Acknowledgements
This work was supported by the NSFC (21207079, 21377067 and 21307062).Notes and references
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- Z. Zou, J. Ye, K. Sayama and H. Arakawa, Nature, 2001, 414, 625 CrossRef CAS PubMed.
- J. Li, Y. Yu and L. Zhang, Nanoscale, 2014, 6, 8473 RSC.
- H. F. Cheng, B. B. Huang and Y. Dai, Nanoscale, 2014, 6, 2009 RSC.
- S. L. Wang, W. H. Ma, Y. F. Fang, M. K. Jia and Y. P. Huang, Appl. Catal., B, 2014, 150–151, 380 CrossRef CAS PubMed.
- J. Wang, Y. Zhang, L. Tian, F. Liu and Q. Xia, J. Nanopart. Res., 2014, 16, 2691 CrossRef.
- Y. F. Fang, W. H. Ma, Y. P. Huang and G. W. Cheng, Chem. Eng. J., 2013, 19, 3224 CAS.
- S. L. Wang, L. L. Wang, W. H. Ma, D. M. Johnson, Y. F. Fang, M. K. Jia and Y. P. Huang, Chem. Eng. J., 2015, 259, 410 CrossRef CAS PubMed.
- G. Absalan, M. Akhond and L. Sheikhian, Amino Acids, 2010, 39, 167 CrossRef CAS PubMed.
- J. T. Edward, P. G. Farrell and J. L. Job, J. Am. Chem. Soc., 1974, 96, 902 CrossRef CAS.
- K. Tada, M. Tada and Y. Maita, J. Oceanogr., 1998, 54, 313 CrossRef CAS.
- N. O. G. Jørgensen, Limnol. Oceanogr., 1987, 32, 97 CrossRef.
- C. Goeschen, N. Wibowo and J. M. White, Org. Biomol. Chem., 2011, 9, 3380 CAS.
- D. P. Li, C. Y. Hu, Y. L. Lin and S. J. Xia, Sci. Total Environ., 2011, 409, 1116 CrossRef PubMed.
- T. Bond, J. Huang, M. R. Templeton and N. Graham, Water Res., 2011, 45, 4321 CrossRef PubMed.
- H. C. Hong, M. H. Wong and Y. Liang, Arch. Environ. Contam. Toxicol., 2009, 56, 638 CrossRef CAS PubMed.
- T. Bond, N. H. M. Kamal and T. Bonnisseau, J. Hazard. Mater., 2014, 278, 288 CrossRef CAS PubMed.
- K. J. Ullrich, G. Rumrich, Th. Wieland and W. Dekant, Pflugers Arch., EJP, 1989, 415, 342 CrossRef CAS.
- A. Campos and V. Vasconcelos, Int. J. Mol. Sci., 2010, 11, 268–287 CrossRef CAS PubMed.
- C. Drahl, B. F. Cravattn and E. J. Sorensen, Angew. Chem., Int. Ed., 2005, 44, 5788 CrossRef CAS PubMed.
- A. Moshnikova, V. Moshnikova, O. A. Andreev and Y. K. Reshetnyak, Biochemistry, 2013, 52, 1171 CrossRef CAS PubMed.
- Y. F. Fang, Y. P. Huang, J. Yang, P. Wang and G. W. Cheng, Environ. Sci. Technol., 2011, 45, 1593 CrossRef CAS PubMed.
- S. Horikoshi, N. Serpone, J. Zhao and H. Hidaka, J. Photochem. Photobiol., A, 1998, 118, 123 CrossRef CAS.
- L. Elsellami, F. Vocanson, F. Dappozze, R. Baudot, G. Febvay, M. Rey, A. Houas and C. Guillard, Appl. Catal., B, 2010, 94, 192 CrossRef CAS PubMed.
- M. Matsushita, T. H. Tran, A. Y. Nosaka and Y. Nosaka, Catal. Today, 2007, 120, 240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional figures and table. See DOI: 10.1039/c5ra09528j |
| This journal is © The Royal Society of Chemistry 2015 |