
Importance of polarization assisted/resonance assisted hydrogen bonding interactions and unconventional interactions in crystal formations of five new complexes bearing chelidamic acid through a proton transfer mechanism†
Masoud Mirzaei*a,
Hossein Eshtiagh-Hosseinia,
Mojtaba Shamsipurb,
Mahdi Saeedia,
Mehdi Ardalanib,
Antonio Bauzác,
Joel T. Magued,
Antonio Fronterac and
Morteza Habibibe
aDepartment of Chemistry, Ferdowsi University of Mashhad, Mashhad 917751436, Iran. E-mail: mirzaeesh@um.ac.ir; Fax: +98-051-38796416; Tel: +98-051-38805554
bDepartment of Chemistry, Razi University, Kermanshah, Iran
cDepartament de Química, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), Spain
dDepartment of Chemistry, Tulane University, New Orleans, Louisiana 70118, USA
eDepartment of Chemistry, Arak University, P.O. Box, 38156-879, Arak, Iran
First published on 20th August 2015
Abstract
(H4a-dmpy)[Cr(Hcda)2]·4H2O (1), (H2a-4,6-dmpym)[Cr(Hcda)2]·3H2O·2a-4,6-dmpym (2), [M(Hcda)(H2O)3]·H2O (3) and (4) (M = CoII and NiII, respectively) and (H2a-4mpy)2[FeCl3(Hcda)]·3H2O (5) (H3cda = 4-hydroxypyridine-2,6-dicarboxylate or chelidamic acid; 4a-dmpy = 4-aminodimethylpyridine; 2a-4,6-dmpym = 2-amino-4,6-dimethylpyrimidine; 2a-4mpy = 2-amino-4-methylpyridine) are five new coordination compounds obtained through the same proton transfer mechanism. The chemical formulas and structures for compounds 1, 2 and 5 are determined by means of single crystal X-ray diffraction, elemental analysis and IR spectroscopy. Single crystal X-ray diffraction was used to characterize compounds 3 and 4. From these results, it has been found that 1, 2 and 5 are ionic with protonated amines as cations and chelidamic acid-complexed metals as anions while 3 and 4 are neutral chelidamic acid complexes of CoII and NiII with the same 3D networks in the crystal. The geometries of all these complexes with Hcda are distorted octahedral around the metal center. Chelidamic acid, as a tridentate ligand, adopts its most common coordination mode through one oxygen atom of each carboxylate group and a nitrogen atom of its pyridine ring in all five complexes. Among various types of non-covalent interactions which result in the stabilized MOFs, hydrogen bonds, particularly very strong polarization assisted hydrogen bonding (PAHB) as well as resonance assisted hydrogen bonding (RAHB), appear to dominate. We have also studied other unconventional interactions observed in the solid state of the complexes using high level DFT calculations. Solution studies were performed to fully characterize the new compounds.
1. Introduction
The hydrogen bond is indeed the most important of all directional intermolecular interactions.1 It strongly affects the structure and properties of many molecules participating in this interaction.2 Recently, numerous studies have been increasingly devoted to strong and very strong hydrogen bonds due to the significant role they play in various fields including proton transfer processes, biochemical reactions and enzyme catalysis.2–4 Proton transfer and hydrogen bonding are strongly linked concepts5–7 and, in some cases, the H-bonding interaction is believed to be an initial step of the proton transfer process.8,9 The strength of an H-bond can be increased to a great extent by two effects: σ-bond cooperativity and π-bond cooperativity, leading to the so-called polarization-assisted hydrogen bonding (PAHB)10–16 and resonance-assisted hydrogen bonding (RAHB),2,17–22 respectively. In recent years, as a continuation of studies on dipicolinic acid,23 Mirzaei et al. have been investigating a very similar dicarboxylic acid (chelidamic acid) as an efficient proton donor in the syntheses of proton transfer compounds. This multicarboxylate and N-heterocyclic ligand is of interest due to its wide applicability in several areas like coordination chemistry, medicine, especially HIV investigations, biochemistry, and above all in proton transfer reactions.5,24We have investigated the coordination behavior of this ligand in the presence of four transition metal ions (Cr(III), Co(II), Ni(II), (Fe(III)) and two amines as proton acceptors (4-aminodimethylpyridine and 2-amino-4,6-dimethylpyrimidine) with alteration of one of three initial materials in each proton transfer reaction. Herein, we report the synthesis and X-ray characterization of five new coordination compounds: (H4a-dmpy)[Cr(Hcda)2]·4H2O (1), (H2a-4,6-dmpym)[Cr(Hcda)2]·3H2O(2a-4,6-dmpym) (2), [M(Hcda)(H2O)3]·H2O (M = CoII, (3) NiII (4)) and (H2a-4mpy)2[FeCl3(Hcda)]·3H2O (5) (see Scheme 1). Moreover, in addition to the strong electrostatically assisted H-bonding interactions, other less studied interactions which also contribute to the crystal packing have been energetically analyzed by means of high level DFT calculations. Solution studies were performed to fully characterize the new compounds.
 | ||
| Scheme 1 Compounds 1–5 reported in this work. | ||
2. Experimental
2.1. Chemicals and measurements
Chemicals were purchased from commercial sources and used without further purification. Infrared spectra (4000–600 cm−1) were recorded on a Buck 500 scientific spectrometer as KBr discs. Elemental analyses (C, H and N contents) were carried out on a Thermo Finnigan Flash-1112EA microanalyzer. The X-ray data were collected with a Bruker Smart APEX diffractometer.2.2. Syntheses of compounds 1–5
For 1–4, an aqueous solution of the appropriate amine was added drop-wisely to an aqueous solution of chelidamic acid (H3cda) followed by drop-wisely addition of an aqueous solution of the appropriate hydrated metal nitrate. Details for 5 are given below.Anal. cal. for C21H25CrN4O14: C, 41.38; H, 4.14; N, 9.20. Found: C, 42.54; H, 3.93; N, 9.51%. Selected IR bands (KBr pellet, cm−1): 1370/1652, νs/νas(COO−); 1448, 1568, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), ν(CC).
N), ν(CC).
2.3. Theoretical methods
The energies of all complexes included in this study were computed at the BP86-D3/def2-TZVP level of theory. We have used the crystallographic coordinates for the theoretical analysis of the non-covalent interactions observed in the solid state. This level of theory has been shown useful and reliable for the study of noncovalent interactions like those analyzed herein.25 The calculations have been performed by using the program TURBOMOLE version 6.5.26 For the calculations we have used the BP86 functional with the latest available correction for dispersion (D3).273. Results and discussion
3.1. IR spectra analyses
The IR spectra of 1, 2 and 5 show the expected absorption bands due to the carboxylate ligand, the amine and water. Those in the 3500–3000 cm−1 region correspond to the N–H and O–H stretching vibrations of the amino groups, water molecules and the chelidamic acid ligand of both compounds. The coordination mode of the carboxylate ligand to the metal ion can be determined by the difference between the asymmetric and symmetric carboxylate stretching frequencies (Δ = νas(COO−) − νs(COO−)). Values above 200 cm−1 indicate monodentate coordination, while those of approximately 164 cm−1 and 42 cm−1 represent ionic and bidentate modes, respectively.23,24 The calculated values for 1 (Δ = 1652 − 1370 = 282 cm−1) and 2 (Δ = 1671 − 1359 = 312 cm−1) imply monodentate coordination for all carboxylate ligands.3.2 Synthesis, characterizations and structural comparison of 1 and 2
From the reaction of the dicarboxylic acid (H3cda) with two equivalents of an amine, 4a-dmpy and 2a-4,6-dmpym, in 1 and 2, respectively, the corresponding proton transfer compounds were obtained and on subsequent addition of aqueous solutions of Cr(III) nitrate compounds 1 and 2 were obtained through slow evaporation of the final solutions. From single crystal X-ray diffraction, elemental analysis and IR spectroscopy the products were characterized as (H4a-dmpy)2[Cr(Hcda)2]·4H2O and (H2a-4,6-dmpym)[Cr(Hcda)2]·3H2O(2a-4,6-dmpym), respectively.| 1 | 2 | |
|---|---|---|
| Empirical formula | C21H25CrN4O14 | C26H31CrN8O13 |
| Formula weight | 609.45 | 715.59 |
| Temperature | 100(2) K | 100(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Orthorhombic |
| Space group | P21 | Pna21 |
| Unit cell dimensions | ||
| a | 15.107(3) Å | 18.7610(18) Å |
| b | 8.9460(17) Å | 8.3037(8) Å |
| c | 19.608(4) Å | 18.9883(19) Å |
| α | 90.00° | 90.00° |
| β | 105.171(3)° | 90.00° |
| γ | 90.00° | 90.00° |
| Volume | 2552.8(8) Å3 | 2958.1(5) Å3 |
| Z | 4 | 4 |
| Density (calculated) | 1.586 mg m−3 | 1.607 mg m−3 |
| Absorption coefficient | 0.528 mm−1 | 0.470 mm−1 |
| F(000) | 1260.0 | 1484.0 |
| Crystal size | 0.23 × 0.19 × 0.10 mm3 | 0.27 × 0.16 × 0.04 mm3 |
| Theta range for data collection | 2.52° to 29.1° | 2.15° to 28.28° |
| Reflections collected | 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 440 |
24049 |
| Independent reflections | 13194[R(int) = 0.0490] |
6807[R(int) = 0.0533] |
| Refinement method | Least-squares matrix: full on F2 | Least-squares matrix: full on F2 |
| Data/restraints/parameters | 13194/1/733 |
6807/1/437 |
| Goodness-of-fit on F2 | 1.029 | 1.019 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0347, wR2 = 0.0818 | R1 = 0.0440, wR2 = 0.1088 |
| R Indices (all data) | R1 = 0.0394, wR2 = 0.0860 | R1 = 0.0503, wR2 = 0.1146 |
| Largest diff. peak and hole | 0.41 and −0.38 e Å−3 | 1.29 and −0.54 e Å−3 |
| Flack parameter | −0.007(6) | 0.017(12) |
 | ||
| Fig. 1 Perspective view of the asymmetric unit of 1, (a); and 2, (b) displacement ellipsoids are drawn at the 50% probability level. | ||
In 1 there are two independent formula units in the asymmetric unit which have very similar geometries while in 2, a molecule of the added amine has co-crystallized with the ionic compound.
In compound 1, Cr1 and Cr2 are coordinated by four carboxylate oxygen atoms (O1, O3, O6 and O8 for Cr1 and O11, O13, O16 and O18 for Cr2) and two nitrogen atoms (N1 and N2 for Cr1 and N3 and N4 for Cr2) from two dianionic Hcda2− ligands. The axial sites on Cr1 and Cr2 are occupied by nitrogen atoms, N1, N2, N3, and N4, with Cr–N bond lengths of 1.967(2), 1.969(2), 1.970(2) and 1.954(2) Å, respectively. The equatorial positions are occupied by four O atoms, with Cr1–O distances of 2.006(2), 1.9841(19), 2.001(2), 2.001(2) Å and Cr2–O bond lengths of 1.986(2), 2.023(2), 2.007(2), 1.983(2) Å for O1, O3, O6, O8 and O11, O13, O16, and O18, respectively.
In compound 2, as for 1, the axial sites are occupied by N1 and N2 with Cr1–N1 and Cr1–N2 bond lengths of 1.964(2) and 1.955(2) Å while O(1), O(3), O(6) and O(8) of the carboxylate ligands occupy the equatorial sites with short Cr–O distances of 1.997(3), 1.988(3), 1.977(3), and 1.986(3) Å, respectively. The bond angles about Cr differ from 90° and indicate distortions of the complexes from strict octahedral geometry (see Table S1†).
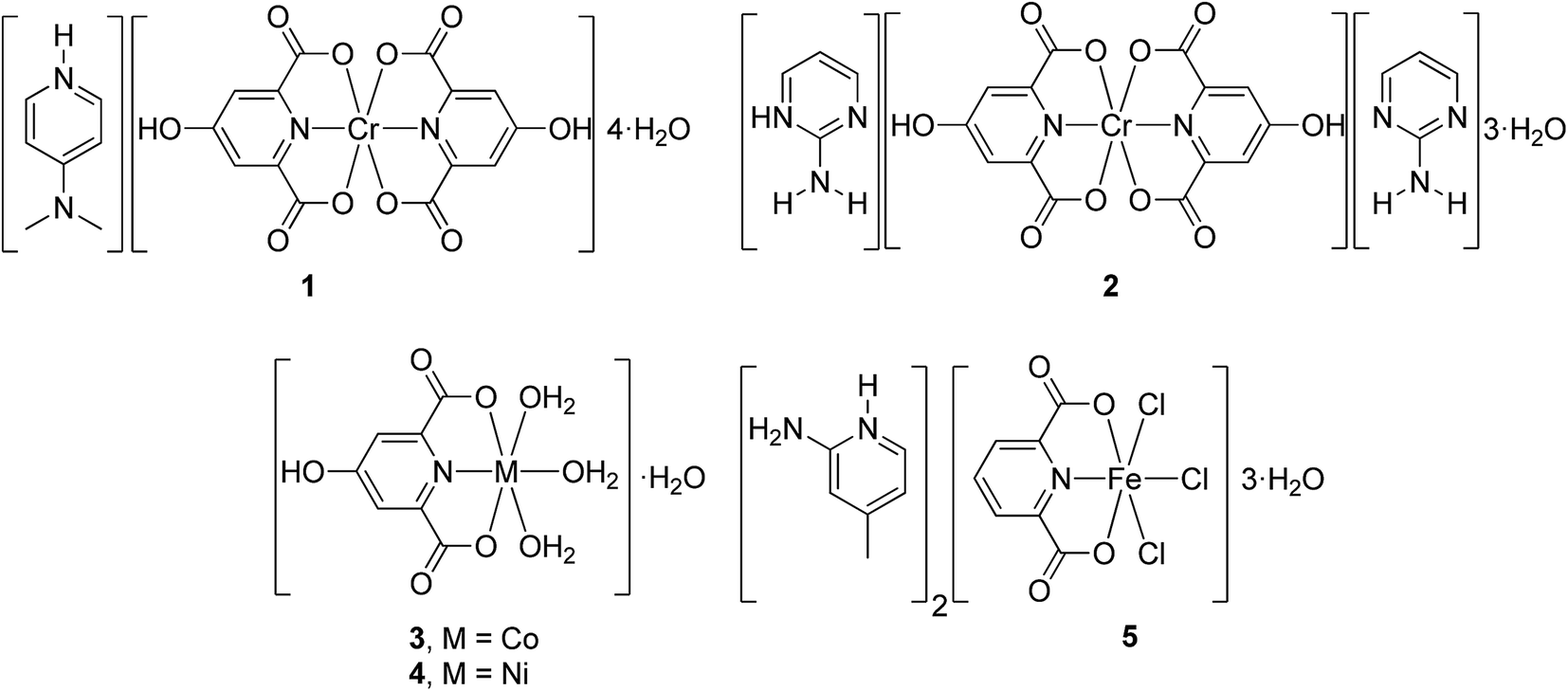
Amongst the several types of non-covalent interactions which are present in the crystals of 1 and 2, very strong hydrogen bonds, particularly, resonance-assisted as well as polarization-assisted hydrogen bonds, seem to be the most significant. These interactions are illustrated in Fig. 2a and b for 1 and 2, respectively.
 | ||
| Fig. 2 Examples of RAHBs and PAHBs, in which each H2O molecule forms up to four (at least three) hydrogen bonds simultaneously, in 1 (a) and 2 (b). | ||
Water molecules as the solvent of both compounds are very effective in mediating the crystal growth and also the stability of the final structures due to their capability of forming up to four hydrogen bonds, two of which are through two lone pairs of electrons on their O atoms as proton acceptors as well as two other H-bonds via two H atoms as donor sites (see Fig. 2). These characteristics are vital for a molecule to be able to form PAHBs through σ-bond cooperativity, which means it accepts and donates protons simultaneously so that all the H-bonds formed are strengthened through polarization enhancement of hydrogen bonds.10–16
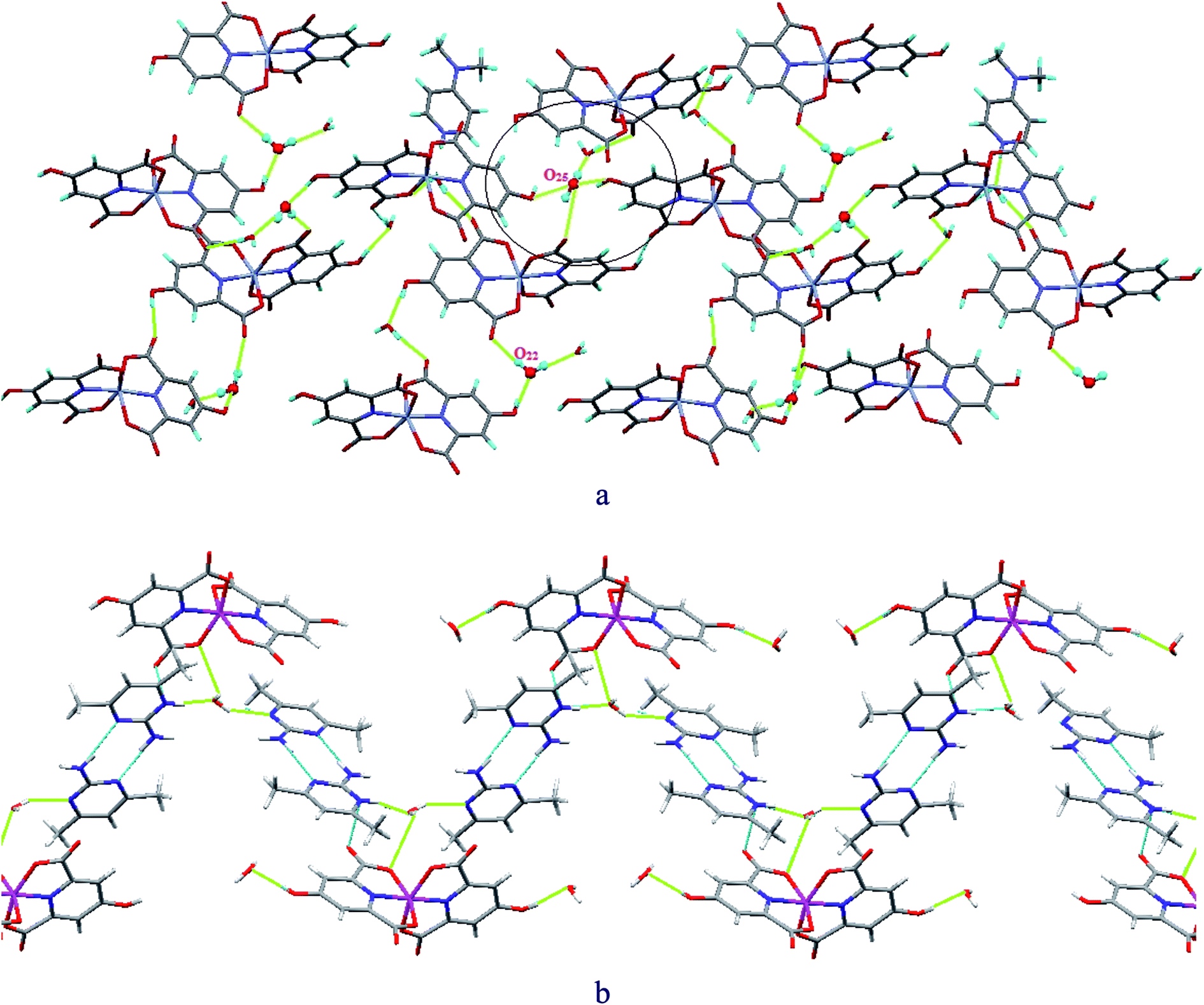
Fig. 3 illustrates several examples of very strong O–H⋯O RAHBs and also PAHBs leading to the formation of R55(10) cyclic motifs and water clusters with H-bonds having extremely short bond lengths as well as D–H⋯A angles close to 180° in compound 1. For instance, O(25)–H(25A)⋯O(23) with a 179° D–H⋯A angle and O(20)–H(20A)⋯O(22) at 1.69 Å represent the bond angle closest to180° and the shortest H-bond length in 1, respectively (see Table S2†).
 | ||
| Fig. 3 Schematic representation of R55(10) cyclic motifs created by very strong O–H⋯O RAHBs and PAHBs in compound 1. | ||
In addition to the hydrogen bonding, other noncovalent interactions, including slipped π–π stacking between pyridine rings and the corresponding adjacent amine molecules (centre–centre distances = 3.536 Å for 1, 3.866 and 3.748 Å for 2, see Fig. 4) also cooperate to create the stabilized 3D supramolecular structures for both compounds. The relevant distances and angles associated with these interactions between the pyridine rings of the chelidamic acid and the pyridine and pyrimidine rings of 4a-dmpy and 2a-4,6-dmpym in 1 and 2 are presented in Fig. 4a and b, respectively. The importance of π–π stacking interactions in crystal growth has been recently studied by Zaric's group.28
 | ||
| Fig. 4 π–π stacking interactions between the pyridine and pyrimidine rings of the cations with the Hcda ligands in 1 and 2, respectively. | ||
The presence of a neutral base or acid molecule in the ultimate structure of compounds formed through proton transfer reactions is not a common phenomenon. Our suggestion for the process of crystal growth provides a rationale for the presence of the free base in the asymmetric unit of 2. Very strong homonuclear N–H⋯N RAHBs make neutral 2a-4,6-dmpym molecules bind to 2-amino-4,6-dimethylpyrimidinum cations to form R22(8) heterosynthons. It is illustrated in Fig. 5 how π-bond cooperativity (resonance due to π-electron delocalization) in the conjugated system leads to the formation of these strong RAHBs.2,17–22
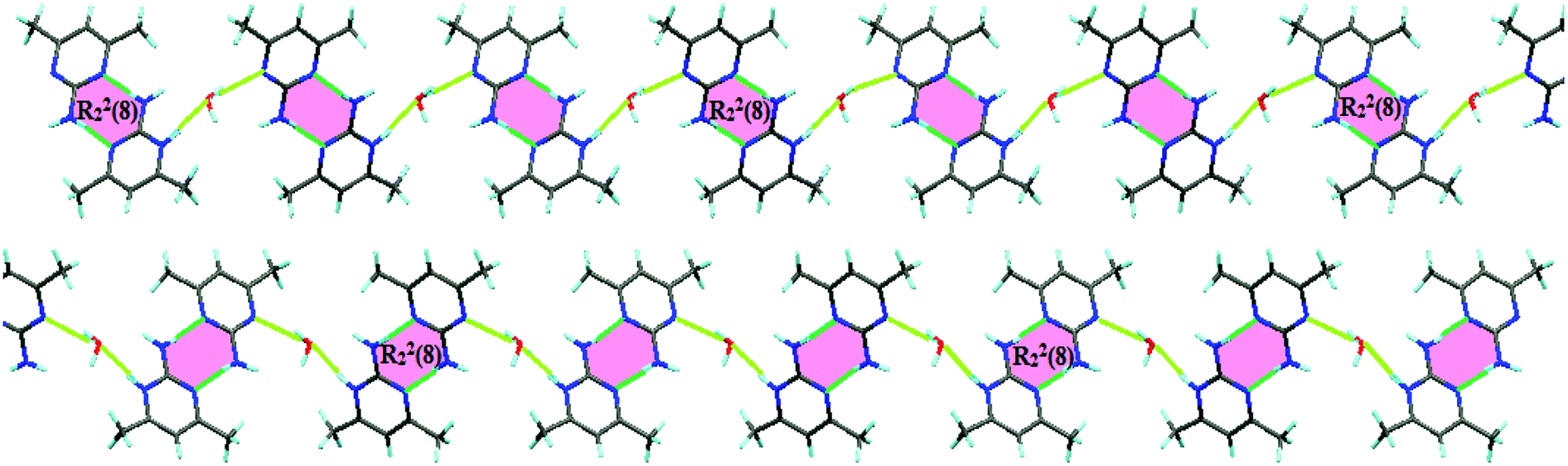 | ||
| Fig. 5 R22(8) Heterosynthons generated by very strong RAHBs between 2-amino-4,6-dimethylpyrimidinum cations and neutral 2a-4,6-dmpym molecules leading to the presence of an unprotonated amine in the formation of crystals of 2. | ||
These units are then linked together by very strong so-called polarization assisted H-bonds formed through σ-bond cooperativity with the help of solvent water molecules, which highlights the aforementioned effectiveness of water molecules in directing the crystal growth (see Fig. 2 and 3).
3.3. Syntheses and crystal structure description of 3 and 4
The crystallographic data for compounds 3 and 4 are shown in Table 2. In addition selected bond lengths, bond angles and hydrogen bond geometries are given in Tables S1 and S2.† Both have the formula [M(Hcda)(H2O)3]·H2O (M = CoII (3), NiII (4)) and are essentially isostructural. Fig. 6 illustrates the asymmetric units with the atomic numbering schemes of 3 and 4, respectively, which consist of an octahedral complex of the corresponding metal ion with three coordinated plus one uncoordinated solvent water molecules (Table 2).| 3 | 4 | |
|---|---|---|
| Empirical formula | C7H9CoNO8·H2O | C7H9NNiO8·H2O |
| Formula weight | 312.10 | 311.88 |
| Temperature | 150 K | 150 K |
| Wavelength | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c |
| Unit cell dimensions | ||
| a | 14.7110(7) Å | 14.7013(6) Å |
| b | 6.8772(3) Å | 6.8433(3) Å |
| c | 22.4185(8) Å | 22.2874(10) Å |
| α | 90.00° | 90.00° |
| β | 90.388(1)° | 90.163(1)° |
| γ | 90.00° | 90.00° |
| Volume | 2268.04(17) Å3 | 2242.22(17) Å3 |
| Z | 8 | 8 |
| Density (calculated) | 1.828 mg m−3 | 1.836 mg m−3 |
| F(000) | 1272 | 1264 |
| Crystal size | 0.22 × 0.13 × 0.09 mm3 | 0.28 × 0.15 × 0.11 mm3 |
| Reflections collected | 20119 |
19591 |
| Refinement method | Least-squares matrix: full on F2 | Least-squares matrix: full on F2 |
| Largest diff. peak and hole | 1.41 and −0.64 e Å−3 | 1.82 and −0.68 e Å−3 |
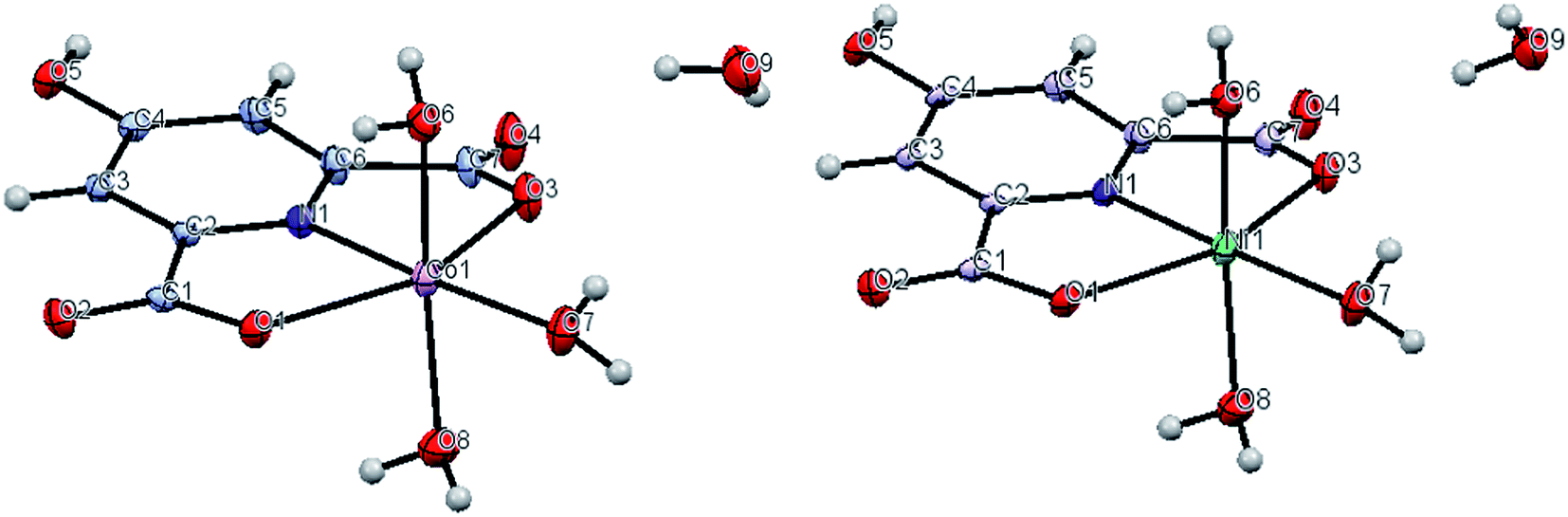 | ||
| Fig. 6 Perspective view of the asymmetric unit of 3 (left) and 4 (right). Displacement ellipsoids are drawn at the 50% probability level. | ||
| 5 | |
|---|---|
| Empirical formula | C19H23Cl3FeN5O5 |
| Formula weight | 563.62 |
| Temperature (K) | 100(2) |
| Wavelength (Å) | 0.71073 |
| Crystal system | Triclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions (Å, °) | |
| a | 10.0736(9) |
| b | 11.177(1) |
| c | 11.6302(11) |
| α | 87.916(1) |
| β | 79.547(1) |
| γ | 64.073(1) |
| Volume (Å3) | 1156.77(18) |
| Z | 2 |
| Density, calc. (g cm−1) | 1.618 |
| F(000) | 578 |
| Crystal size (mm) | 0.150 × 0.150 × 0.230 |
| Reflections collected | 20506 |
| Refinement method | Least-squares matrix: full on F2 |
| Largest diff. peak and hole (e Å−3) | 0.42 and −0.49 |
Unlike 1 and 2, the complexes here are neutral species and include only one (Hcda)2− ligand bound to the central transition metal ion in its most common coordination mode as a tridentate chelate (two carboxylate oxygen atoms, O1 and O3 and the nitrogen atom, N1). The remaining three sites on the metal ions are occupied by H2O molecules (O6, O7 and O8). There is one uncoordinated H2O (H9A–O9–H9B) in both compounds as well. This solvent water molecule plays a significant role in the formation of non-covalent interactions and subsequently in the stabilization of the crystal structure (see Fig. 7).
 | ||
| Fig. 7 Lattice water molecules forming O–H⋯O PAHBs in 3 and 4. | ||
Recently, Mirzaei et al. reported dipicolinic acid compounds of CoII and NiII (ref. 23) prepared by reactions analogous to those used for 3 and 4 which are also neutral species so that the reactant bases are not incorporated into the crystal structures. Here, however, the complexes contain two mono-deprotonated dipicolinic acid ligands which function as tridentate chelates and while it is not obvious why the two systems behave differently, it is tempting to suggest that the additional hydrogen bonding available in chelidamic acid because of the additional hydroxyl group may be important.
In addition to hydrogen bonding, slipped π–π stacking interactions between pyridine rings of the chelidamate ligands in the complexes (centre–centre distances = 4.086 Å for 3, 4.077 Å for 4, see Fig. 8) are effective as well to create the stabilized MOFs for both compounds. The corresponding rings distances and angles between pyrimidine rings are presented in Fig. 4.
 | ||
| Fig. 8 π–π stacking interactions between pyridine rings of chelidamate ligands in complexes 3 (left) and 4 (right). | ||
The coordination geometry of iron in 5 is distorted octahedral with the dianionic (Hcda)2− ligand coordinated as in the previous complexes and the remaining sites occupied by Cl ligands. The 2-amino-4-methyl pyridinium cations are protonated on the ring nitrogen as expected and engage in extensive hydrogen bonding interactions with the anions and the lattice water to generate a layer structure (Table 3). Moreover, in 5 it is interestingly observed that the compound is composed of an anionic complex of Fe(III), [FeCl3(Hcda)]−2, having three sites of the metal ion occupied by three chloride ions. The compound, which is neutralized by the two positively-charged amine ligands shows the same moral ratio as that of adopted for initial materials.
3.4. Theoretical study
We have analyzed the energetics of the noncovalent interactions observed in the solid state of compounds 1, 2 and 5 focusing our attention on the lone pair (lp)–π, π-hole and antiparallel C–O⋯C–O interactions. We have selected these compounds because they present unconventional and less studied interactions in the solid state that are worthy of investigation. Compounds 3 and 4 present more conventional H-bonding and π-stacking interactions, and so have not been included here.Recently, π-hole interactions have attracted attention in supramolecular chemistry and crystal engineering fields.29 In the crystal structure of compound 1 we have observed that the oxygen atom of a lattice water molecule is close to the carbon atom of one carboxylate group coordinated to the metal center (see Fig. 9A) and the distance, 3.098(4) Å, is significantly shorter than the sum of the van der Waals radii (3.22 Å). We have computed two theoretical models (C and D in Fig. 9) where one cation (protonated 4-aminodimethylpyridine) has been included in order to use models that include both counterions and the water molecule. The interaction energy of model C is ΔE1 = −19.9 kcal mol−1 that corresponds to the contribution of two strong H-bonds (denoted as HB1 and HB2 in Fig. 9) and the O → C π-hole interaction. In model D we have substituted the carboxylate ligand that participates in the O → C π-hole interaction with a hydrogen and added a hydrogen atom to the aromatic ring (see arrows in Fig. 9D). In this model, only the contribution of the H-bonding interactions are evaluated. The resulting interaction energy for this model is reduced to ΔE2 = −17.6 kcal mol−1. Therefore the contribution of the lp–π-hole interaction is ΔE1 − ΔE2 = −2.3 kcal mol−1. The existence of the π-hole has been confirmed by means of the Molecular Electrostatic Potential Surface (MEPS) that is shown in Fig. 9B. It can be observed as a region (light blue) just over the carbon atom of the coordinated carboxylate group.
 | ||
| Fig. 9 (A) A fragment of the experimental crystal structure of compound 1. (B) MEP surface computed for a model of 1. (C and D) Theoretical model used to evaluate the π-hole interaction. (E) AIM distribution of bond (red spheres) and ring (yellow spheres) critical points. The bond paths are also represented. | ||
In complex 4 we have studied theoretically the antiparallel C–O⋯C–O interaction observed in the phenolic part of the ligand. It has an important role in the crystal packing of 4 (see Fig. 10A). This type of interaction has been described for carbonyl compounds (CO⋯CO interaction),30 but to our knowledge it has not been studied for phenolic compounds. The theoretical models to analyze the energetics of the C–O⋯C–O interaction are shown in Fig. 10. The interaction energy of the self-assembled dimer (Fig. 10B) is ΔE3 = −3.1 kcal mol−1 which is comparable to that previously described for carbonyl compounds.30 We have also studied the effect of the metal coordination on the binding strength by computing an additional theoretical model where the metal centers have been eliminated and the carboxylate groups have been protonated (see Fig. 10C). As a result, the interaction energy is similar (ΔE3 = −3.6 kcal mol−1) indicating that the metal coordination does not influence the binding strength. The MEP surface computed for compound 4 shows that the electrostatic potential over the O atom is slightly negative (−2.0 kcal mol−1) and positive over the C atom, thus favoring the antiparallel C–O⋯C–O interaction.
 | ||
| Fig. 10 (A) A fragment of the experimental crystal structure of compound 4. (B and C) Theoretical model used to evaluate the antiparallel C–O⋯C–O interaction. (D) MEP surface computed for a model of 4. (E) AIM distribution of bond (red spheres) and ring (yellow spheres) critical points. The bond paths are also represented. | ||
Finally, in complex 5, we have analyzed the stacking interaction between two protonated aminopyridine rings that has an important role in the solid state architecture of 5. This stacking interaction is antiparallel and it has some peculiarities. First, the lone pair of the amino group of one moiety interacts with the pyridine ring of the other and vice versa (resembling a double lp–π interaction, see red dashed lines in Fig. 11A). Second, the interaction is established between two cationic moieties (protonated pyridine); therefore a strong electrostatic repulsion exists disfavoring the interaction. However, each dianionic [FeCl3(Hcda)]2− moiety establishes a double salt bridge interaction with two aminopyridine rings (see Fig. 11A) and therefore most of the positive charge is transferred to the counterion. In addition, if the interaction is considered as an lp–π type, it is enhanced by the protonated pyridine ring. Using the theoretical models shown in Fig. 11 we have evaluated the double lp–π interaction. First we have used the (H2a-4mpy)2[FeCl3(Hcda)] salt as one part of the complex and the protonated aminopyridine as the other part. As a result the interaction energy is very large and negative, indicating that it is very favorable (ΔE5 = −25.8 kcal mol−1). This strong interaction is also due to purely electrostatic ion-pair interaction with the anionic complex. In fact, if the interaction is computed using model C where only neutral aminopyridine rings are considered, the interaction is drastically reduced to ΔE6 = −6.3 kcal mol−1. This is further confirmed by the MEPS analysis, which shows a positive potential over the center of the aromatic ring and a negative one over the lone pair of the amino group (see Fig. 11E).
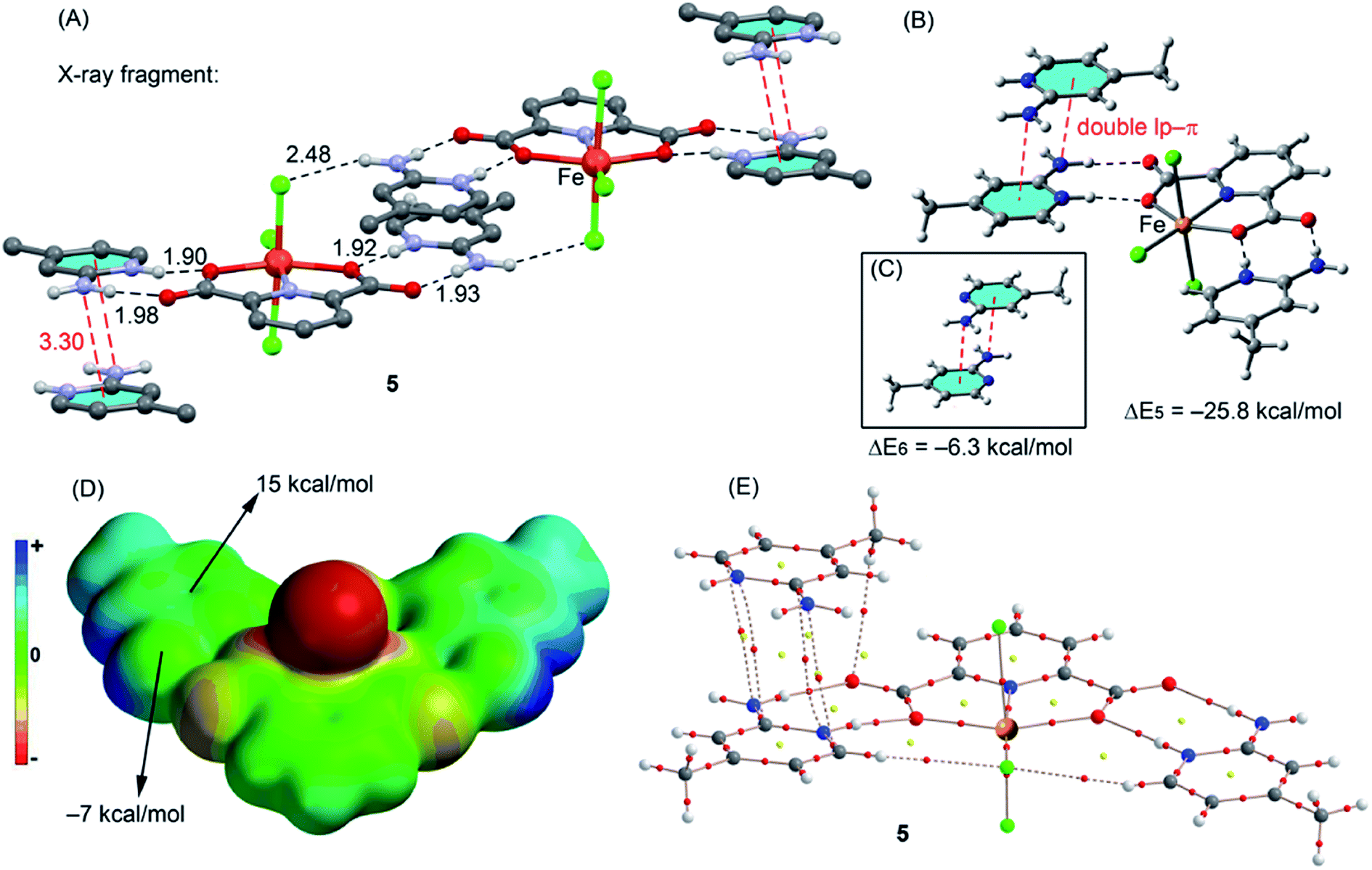 | ||
| Fig. 11 (A) A fragment of the experimental crystal structure of compound 5. (B and C) Theoretical model used to evaluate the double lp–π interaction. (D) MEP surface computed for a model of 5. (E) AIM distribution of bond (red spheres) and ring (yellow spheres) critical points. The bond paths are also represented. | ||
We have used Bader's “atoms-in-molecules” approach that has been successfully used to describe a great variety of noncovalent interactions to further analyze the interactions described above for complexes 1, 4 and 5. The presence of a bond critical point (CP) and a bond path connecting two atoms is an unambiguous indication of interaction.31 The distribution of critical points in compound 1 (Fig. 9E) reveals the presence of a bond CP that connects the O atom of the water molecule with the carbon atom of the carboxylate confirming the π-hole interaction. Furthermore, the distribution of CPs also reveals the presence of a bond CP connecting the H atom of water with the apical oxygen atom of the carboxylate that characterizes the H-bonding interaction. In compound 4 (Fig. 10E) the antiparallel C–O⋯C–O interaction is characterized by the presence of two bond CPs that connect the C atoms to the O atoms. The interaction is further characterized by the presence of a ring CP as a consequence of the formation of the supramolecular four membered ring. Finally, in compound 5 (Fig. 11E) the double lp–π interaction is confirmed since there are two bond CPs that connect each N atom of the amino group to one carbon atoms of the pyridine ring. Moreover, the complex is also characterized by the presence of two bond CPs connecting both aromatic rings.
3.5. Solution studies
In order to determine the stoichiometry and stability of the Cr3+, Co2+ and Ni2+ (M) complexes with hypydc (L), 4a-dmpy (Q) and hypydc-4a-dmpy in aqueous solution, a 2.4 × 10−3 M solution of L, a 4.8 × 10−3 M solution of Q and their mixture, all containing a necessary amount of 0.85 M HCl, were titrated in the absence and presence of 1.2 × 10−3 M of the metal ion with a 0.096 M solution of NaOH at a temperature of 25 °C and ionic strength of 0.1 M, maintained by KNO3. The corresponding equilibrium potentiometric pH titration profiles are shown in Fig. 12a–c, respectively. The pH titration data in the absence of metal ions were used to calculate the protonation constants for L and Q (Knh[HmL]/[H(m–n)L][H]n, the charges are omitted for simplicity) via the program BEST.31 The resulting values for the overall stability and stepwise protonation constants of L and Q as well as the recognition constants for the L–Q proton transfer system are listed in Table 4. As it is obvious from Table 4, the most abundant proton transfer species present at pH 2.0 with an extent of 98.7% is hypydcH2-4a-dmpyH (logK = 13.18). In all three cases the potentiometric titration curves are depressed considerably in the presence of the metal ions, clearly indicating strong interactions of L with the metal ions.
 | ||
| Fig. 12 Potentiometric titration curves of hypydc (a) and 4a-dmpy (b) and their mixture (c) in the absence and presence of Mn+ ions with NaOH 0.096 M in aqueous solution at 25 °C μ = 0.1 M of KNO3. | ||
| Stoichiometry | logβ |
Equilibrium quotient K | logK |
Max% | At pH | ||
|---|---|---|---|---|---|---|---|
| hypydc | 4a-dmpy | h | |||||
| 1 | 0 | 1 | 10.41 | — | 10.41 | 99.9 | 6–7.3 |
| 1 | 0 | 2 | 13.35 | — | 2.94 | 56.5 | 2.5 |
| 1 | 0 | 3 | 15.46 | — | 2.11 | 53.6 | 2 |
| 0 | 1 | 1 | 10.07 | — | 10.07 | 99.9 | 4.2–7 |
| 0 | 1 | 2 | 11.18 | — | 1.11 | 11.4 | 2 |
| 1 | 2 | 0 | 12.97 | [hypydc(4a-dmpy)2]/[hypydc][4a-dmpy]2 | — | 99.9 | 11.3–12 |
| 1 | 2 | 2 | 32.70 | [hypydcH2(4a-dmpy)2]/[hypydcH2][4a-dmpy]2 | 11.88 | 99.9 | 6.9–8.3 |
| 1 | 2 | 3 | 36.60 | [hypydcH2(4a-dmpyH)(4a-dmpy)]/[hypydcH2][4a-dmpyH][4a-dmpy] | 13.18 | 98.7 | 2 |
The corresponding distribution diagrams for hypydc (a), 4a-dmpy (b), hypydc/4a-dmpy (c) and hypydc/Cr3+ (d) and other systems are also shown in Fig. 13 (see Table 5). In the case of Cr3+ (M) with hypydc (L) in the binary system, the most abundant species are ML2(OH)3 existing at pH 10.5–12 by an extent of 99.8%, ML2 existing at pH 4.5–4.8 by an extent of 99.6% and ML2(OH)2 existing at pH 6.8 by an extent of 86.4%. In the case of Cr3+ with 4a-dmpy (Q) in the binary system, the most abundant species are MQ2(OH)3 existing at pH 10.9–12 by an extent of 88.9% and its protonated form MQ2H2 at pH 2.0 by an extent of 88.2%. In the case of Cr3+ with L and Q in the ternary complexes system, the most abundant species are MLQ2(OH)3 existing at pH 9.6–12 by an extent of 88.1%, MLQ2H3 existing at pH 2.4 by an extent of 86.2% and MLQ2H2 existing at pH 4.9 by an extent of 59.0%.
 | ||
| Fig. 13 Distribution diagrams for hypydc (a), 4a-dmpy (b), hypydc/4a-dmpy (c), hypydc/Cr3+ (d), 4a-dmpy/Cr3+ (e), hypydc/4a-dmpy/Cr3+ (f), hypydc/Co2+ (g), hypydc/Ni2+ (h). | ||
| System | m | l | q | h | logβ |
Max% | At pH |
|---|---|---|---|---|---|---|---|
| Cr–hypydc | 1 | 2 | 0 | 0 | 16.92 | 99.6 | 4.5–4.8 |
| 1 | 2 | 0 | 1 | 17.47 | Negligible | — | |
| 1 | 2 | 0 | −2 | 4.64 | 86.4 | 6.8 | |
| 1 | 2 | 0 | −3 | −3.13 | 99.8 | 10.5–12 | |
| Cr–4a-dmpy | 1 | 0 | 2 | 1 | 22.53 | 81.6 | 5.1–5.4 |
| 1 | 0 | 2 | 2 | 26.57 | 88.2 | 2 | |
| 1 | 0 | 2 | −1 | 10.47 | 50.8 | 6.3 | |
| 1 | 0 | 2 | −2 | 3.84 | 76 | 7.6–7.8 | |
| 1 | 0 | 2 | −3 | −4.94 | 88.9 | 10.9–12 | |
| Cr–hypydc–4a-dmpy | 1 | 1 | 1 | 3 | 31.83 | Negligible | — |
| 1 | 1 | 2 | 0 | 24.62 | 12.8 | 6.2 | |
| 1 | 1 | 2 | 1 | 31.22 | 47.5 | 5.9 | |
| 1 | 1 | 2 | 2 | 36.78 | 59 | 4.9 | |
| 1 | 1 | 2 | 3 | 41.11 | 86.2 | 2.4 | |
| 1 | 1 | 2 | −1 | 18.47 | 16.4 | 6.4 | |
| 1 | 1 | 2 | −2 | 12.29 | 32.3 | 6.6 | |
| 1 | 1 | 2 | −3 | 5.71 | 88.1 | 9.6–12 | |
| Co–hypydc | 1 | 2 | 0 | 0 | 17.66 | 32.6 | 6.3 |
| 1 | 2 | 0 | 1 | 23.98 | 69.4 | 5.6 | |
| 1 | 2 | 0 | 2 | 28.91 | 81 | 4 | |
| 1 | 2 | 0 | 3 | 31.97 | 91.8 | 2 | |
| 1 | 2 | 0 | −1 | 11.34 | 73.6 | 7.2 | |
| 1 | 2 | 0 | −2 | 3.44 | 96.8 | 9.5–9.8 | |
| 1 | 2 | 0 | −3 | −8.04 | 72.2 | ≥11.9 | |
| Ni–hypydc | 1 | 2 | 0 | 0 | 14.99 | 68.8 | 6.6 |
| 1 | 2 | 0 | 1 | 20.76 | 26 | 5.9 | |
| 1 | 2 | 0 | 2 | 26.73 | 50.4 | 5.4 | |
| 1 | 2 | 0 | 3 | 31.91 | 99.8 | 2–2.7 | |
| 1 | 2 | 0 | −1 | 7.81 | 98.4 | 9.1–9.5 | |
| 1 | 2 | 0 | −2 | −3.57 | 76.8 | ≥11.9 |
4. Conclusions
We have synthesized and characterized by X-ray diffraction five coordination compounds of 4-hydroxypyridine-2,6-dicarboxylate which coordinates to the transition metals Cr, Co, Ni and Fe as a tridentate ligand. The two anionic Cr(III) complexes contain two nearly perpendicularly chelidamate ligands accompanied by the corresponding protonated amines to form compounds 1 and 2, while the Co(II) and Ni(II), compounds 3 and 4, are neutral complexes having only one chelidamate ligand coordinated to the transition metal ion. Among non-covalent interactions, different types of hydrogen bonding, especially strong resonance- and polarization-assisted ones, which are made through σ bond and π bond cooperativity, followed by stacking interactions are the principal intermolecular interactions in the solid state architectures. Very strong homonuclear N–H⋯N RAHBs make neutral 2a-4,6-dmpym molecules bind to (H2a-4,6-dmpym)+ so that it is part in the asymmetric unit of compound 2. Moreover, the interesting noncovalent interactions observed in the solid state of some compounds have been studied by means of DFT calculations assigning discrete energetic values to them. The most important finding is the antiparallel C–O⋯C–O interaction between the phenolic groups. All interactions have been characterized both energetically and using the “atoms-in-molecules” methodology.Acknowledgements
We thank to the Ferdowsi University of Mashhad for financial support and Tulane University for support of the Tulane Crystallography Laboratory (Grant no. 18972/3). AF and AB thank the MINECO of Spain for financial support (CONSOLIDER-Ingenio 2010 project CSD2010-0065, FEDER funds) and CTI (UIB) for computational facilities.References
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- R. W. Góra, M. Maj and S. J. Grabowski, Phys. Chem. Chem. Phys., 2013, 15, 2514–2522 RSC; S. J. Grabowski, Phys. Chem. Chem. Phys., 2013, 15, 7249–7259 RSC.
- S. J. Grabowski, Chem Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- Hydrogen Bonding – New Insights, in Series Challenges and Advances in Computational Chemistry and Physics, ed. S. J. Grabowski and J. Leszczynski, Springer, New York, 2006 Search PubMed.
- (a) M. Mirzaei, H. Eshtiagh-Hosseini, S. Zarghami, A. Bauza, A. Frontera, J. T. Mague, M. Habibi and M. Shamsipur, CrystEngComm, 2014, 16, 1359 RSC; (b) M. Mirzaei, H. Aghabozorg and H. Eshtiagh-Hosseini, J. Iran. Chem. Soc., 2011, 8, 580 CrossRef CAS; (c) H. Eshtiagh-Hosseini, M. Mirzaei and N. Alfi, Review on Proton Transfer Metal Complexes, Lambert Academic Publishing GmbH & Co. KG., 1st edn, 2012 Search PubMed.
- H. Eshtiagh Hosseini, H. Aghabozorg, M. Shamsipur, M. Mirzaei and M. Ghanbari, J. Iran. Chem. Soc., 2011, 8, 762 CrossRef CAS.
- M. Mirzaei, H. Eshtiagh-Hosseini, M. Chahkandi, N. Alfi, A. Shokrollahi, N. Shokrollahi and A. Janiak, CrystEngComm, 2012, 14, 8468 RSC.
- H.-B. Bürgi, Angew. Chem., 1975, 87, 461 CrossRef.
- H.-B. Bürgi and J. D. Dunitz, Acc. Chem. Res., 1983, 16, 153 CrossRef.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, 1939 Search PubMed.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- G. Gilli and P. J. Gilli, J. Mol. Struct., 2000, 1, 552 Search PubMed.
- G. A. Jeffrey, Crystallogr. Rev., 1995, 3, 213 CrossRef.
- T. Steiner, S. A. Mason and W. Saenger, J. Am. Chem. Soc., 1991, 113, 5676 CrossRef CAS.
- B. M. Kariuki, K. D. M. Harris, D. Philp and J. M. A. Robinson, J. Am. Chem. Soc., 1997, 119, 679 CrossRef.
- S. Scheiner, Hydrogen Bonding, A Theoretical Perspective, Oxford University Press, Oxford, 1997 Search PubMed.
- J. F. Beck and Y. J. Mo, Comput. Chem., 2006, 28, 455 CrossRef PubMed.
- G. Gilli, F. Bellucci, V. Ferretti and V. Bertolasi, J. Am. Chem. Soc., 1989, 111, 1023 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1991, 113, 4917 CrossRef CAS.
- V. Bertolasi, P. Gilli, V. Verretti and G. Gilli, Chem.–Eur. J., 1996, 2, 925 CrossRef CAS.
- C. Bilton, F. H. Allen, G. P. Shields and J. A. K. Howard, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 2000, 56, 849 CrossRef PubMed.
- S. J. Grabowski, J. Mol. Struct., 2001, 562, 137 CrossRef CAS.
- M. Mirzaei, H. Eshtiagh-Hosseini, Z. Karrabi, K. Molčanov, E. Eydizadeh, J. T. Mague, A. Bauza and A. Frontera, CrystEngComm, 2014, 16, 5352 RSC.
- (a) M. Searcey, S. MeClean, B. Madden, A. T. McGown and L. P. G. Wakelin, Anti-Cancer Drug Des., 1998, 13, 837 CAS; (b) D. L. Boger, J. Hong, M. Hikota and M. Ishida, J. Am. Chem. Soc., 1999, 121 Search PubMed.
- (a) L. K. Das, R. M. Kadam, A. Bauzá, A. Frontera and A. Ghosh, Inorg. Chem., 2012, 51, 12407–12418 CrossRef CAS PubMed; (b) M. Mitra, P. Manna, A. Bauzá, P. Ballester, S. Kumar Seth, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, J. Phys. Chem. B, 2014, 118, 14713–14726 CrossRef CAS PubMed; (c) S. Saha, A. Sasmal, C. R. Choudhury, G. Pilet, A. Bauzá, A. Frontera, S. Chakraborty and S. Mitra, Inorg. Chim. Acta, 2015, 425, 211–220 CrossRef CAS; (d) A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed; (e) A. Bauzá, A. Terrón, M. Barceló-Oliver, A. García-Raso and A. Frontera, Inorg. Chim. Acta, 2015 DOI:10.1016/j.ica.2015.04.028; (f) D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 11, 1958–1972 CrossRef; (g) P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- R. Ahlrichs, M. Bär, M. Hacer, H. Horn and C. Kömel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- (a) Z. D. Tomic, D. Sredojevic and S. D. Zaric, Cryst. Growth Des., 2006, 6, 29–31 CrossRef CAS; (b) P. V. Petrović, G. V. Janjić and S. D. Zarić, Cryst. Growth Des., 2014, 14, 3880–3889 CrossRef; (c) D. P. Malenov, D. B. Ninković, D. N. Sredojević and S. D. Zarić, ChemPhysChem, 2014, 15, 2458–2461 CrossRef CAS PubMed; (d) A. Bauza, T. J. Mooibroek and A. Frontera, Chem. Commun., 2015, 51, 1491–1493 RSC; (e) S. Roy, A. Bauza, A. Frontera, R. Banik, A. Purkayastha, M. G. B. Drew, B. M. Reddy, B. Sridhar, S. Kr and D. S. Das, CrystEngComm, 2015, 17, 3912–3916 RSC; (f) P. Pal, S. Konar, M. S. El Fallah, K. Das, A. Bauza, A. Frontera and S. Mukhopadhyay, RSC Adv., 2015, 5, 45082–45091 RSC.
- (a) C.-Q. Wan and T. C. W. Mak, Cryst. Growth Des., 2011, 11, 832–842 CrossRef CAS; (b) M. Barceló-Oliver, B. A. Baquero, A. Bauzá, A. García-Raso, A. Terron, I. Mata, E. Molins and A. Frontera, CrystEngComm, 2012, 14, 5777–5784 RSC; (c) M. Mitra, P. Manna, A. Bauzá, P. Ballester, S. K. Seth, S. R. Choudhury, A. Frontera and S. Mukhopadhyay, J. Phys. Chem. B, 2014, 118, 14713–14726 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- E. Martell and R.-J. Motekaitis, Determination and Use of Stability Constants, 2nd edn, VCH, New York, 1992 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1056388–1056391 (1–4) and 1402098 (5). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09526c |
| This journal is © The Royal Society of Chemistry 2015 |