DOI:
10.1039/C5RA09508E
(Paper)
RSC Adv., 2015,
5, 64566-64581
A facile synthesis and evaluation of new biomolecule-based coumarin–thiazoline hybrids as potent anti-tubercular agents with cytotoxicity, DNA cleavage and X-ray studies†‡
Received
20th May 2015
, Accepted 20th July 2015
First published on 20th July 2015
Abstract
An efficient and rapid synthesis of coumarin–thiazoline hybrids (1a–1j) under microwave irradiation is described with high yields. The synthesized compounds were characterized using elemental and spectroscopic analysis; in addition, the structures of compounds 1a, 1b, 1e and 1h have been elucidated using single crystal X-ray diffraction techniques. All the newly synthesized compounds were screened for their in vitro anti-tubercular activity and in a DNA cleavage study, while the most active compounds were subjected to a cytotoxicity assay on Vero cell lines. Among those tested, compound 1b exhibited excellent anti-tubercular activity (MIC 0.09 μg ml−1) with a low level of cytotoxicity, suggesting that compound 1b is a promising lead for subsequent investigations in search of new anti-tubercular agents. Furthermore, a DNA cleavage study using an agarose gel electrophoresis method revealed that compounds 1b, 1d, 1f and 1i cleaved DNA more efficiently and thereby exhibit nuclease activity.
Introduction
Today, classic drug development works with small, chemically manufactured active substance molecules, since these molecules can reach almost any desired destination in the body and their small structure and chemical composition often helps them to easily penetrate cell membranes and thereby increases the bioavailability of the compound.1 Hence the majority of pharmaceuticals and biologically active drugs are all small-sized molecules. Based on these facts, medicinal chemist Christopher Lipinski and his colleagues, in the year 1997, analyzed the physicochemical properties of more than 2000 drugs and candidate drugs in clinical trials, and concluded that a compound is more likely to be membrane permeable and easily absorbed by the body if it matches the Rule of Five (RO5).2 The rules, based on the 90 percentile values of the analysed drugs’ property distributions, apply only to absorption by passive diffusion of the compound through cell membranes; compounds that are actively transported through cell membranes by transporter proteins are exceptions to the rule. Furthermore, candidate drugs that conform to the RO5 tend to have lower attrition rates during clinical trials and hence have an increased chance of reaching the market.3 Hence, keeping these factors in view the architecture of the compounds was designed and their physicochemical properties (RO5) were analyzed which are listed below.
• All the newly synthesized compounds’ molecular weights fall below the 500 daltons limit.
• The compound’s lipophilicity, expressed as a quantity known as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P, is less than 5 (logP values of all the compounds were calculated using http://www.molinspiration.com) and the values are given in Table 2.
P, is less than 5 (logP values of all the compounds were calculated using http://www.molinspiration.com) and the values are given in Table 2.
• The number of groups in the molecule that can donate hydrogen atoms to hydrogen bonds is less than 5 (expressed as the sum of OHs and NHs).
• The number of groups that can accept hydrogen atoms to form hydrogen bonds is less than 10 (estimated by the sum of Os and Ns).
From the results, it is clear that none of the compounds violate the rules and they fall well within the range as stated by the RO5 to qualify as a drug candidate.
Coumarins are an elite class of oxygen-containing fused heterocycles, which are widely distributed in nature, especially in plants. They form a large class of important lactones with a fused structure of benzene and an α-pyrone ring, and virtually contain a π–π conjugated system rich in electrons and with good charge transport properties. Moreover, the unique structure of coumarin has a special ability which allows its derivatives to readily interact with a diversity of enzymes and receptors in organisms through weak bond interactions and thereby exhibit wide potential as medicinal drugs. Hence, coumarin based compounds have attracted special interest in the area of medicinal chemistry and their outstanding contributions in the prevention and treatment of numerous diseases have become an extremely attractive highlight.4 Recently many coumarin scaffolds have been investigated as potential candidates for the treatment of tuberculosis, e.g. diaryl coumarin (1, Fig. 1), 4-aryl/alkyl sulfonyl methyl coumarin (2), iodinated-4-aryloxymethyl coumarin (3) and chalconated coumarin (4) have been reported to exhibit potent anti-tubercular activity with MICs of 0.24, 0.78, 1.56 and 3.5 μg ml−1 respectively,5 whereas naturally occurring coumarins such as ferulenol (5), suberosin (6), osthol (7), scopoletin (8) and umbelliferone (9) have exhibited MICs of 2, 16, 32, 42 and 58.3 μg ml−1 respectively.6 Reports have also suggested that the coumarin class of compounds targets the fatty acyl-ACP synthetase activity of the FadD32 enzyme5a,6a which is essential for Mtb survival as it plays a critical role in the biosynthesis of the unique branched fatty acids (mycolic acids) that make up the Mtb cell wall. Hence looking into the biological significance of coumarins, particularly in the field of tuberculosis, we anticipate that coumarins could be a good starting point for the development of new lead anti-tubercular drugs. Fig. 1 represents the structures of some potent coumarin scaffolds exhibiting anti-tubercular properties.
 |
| | Fig. 1 Synthetic and natural occurring coumarin derivatives exhibiting anti-tubercular properties. | |
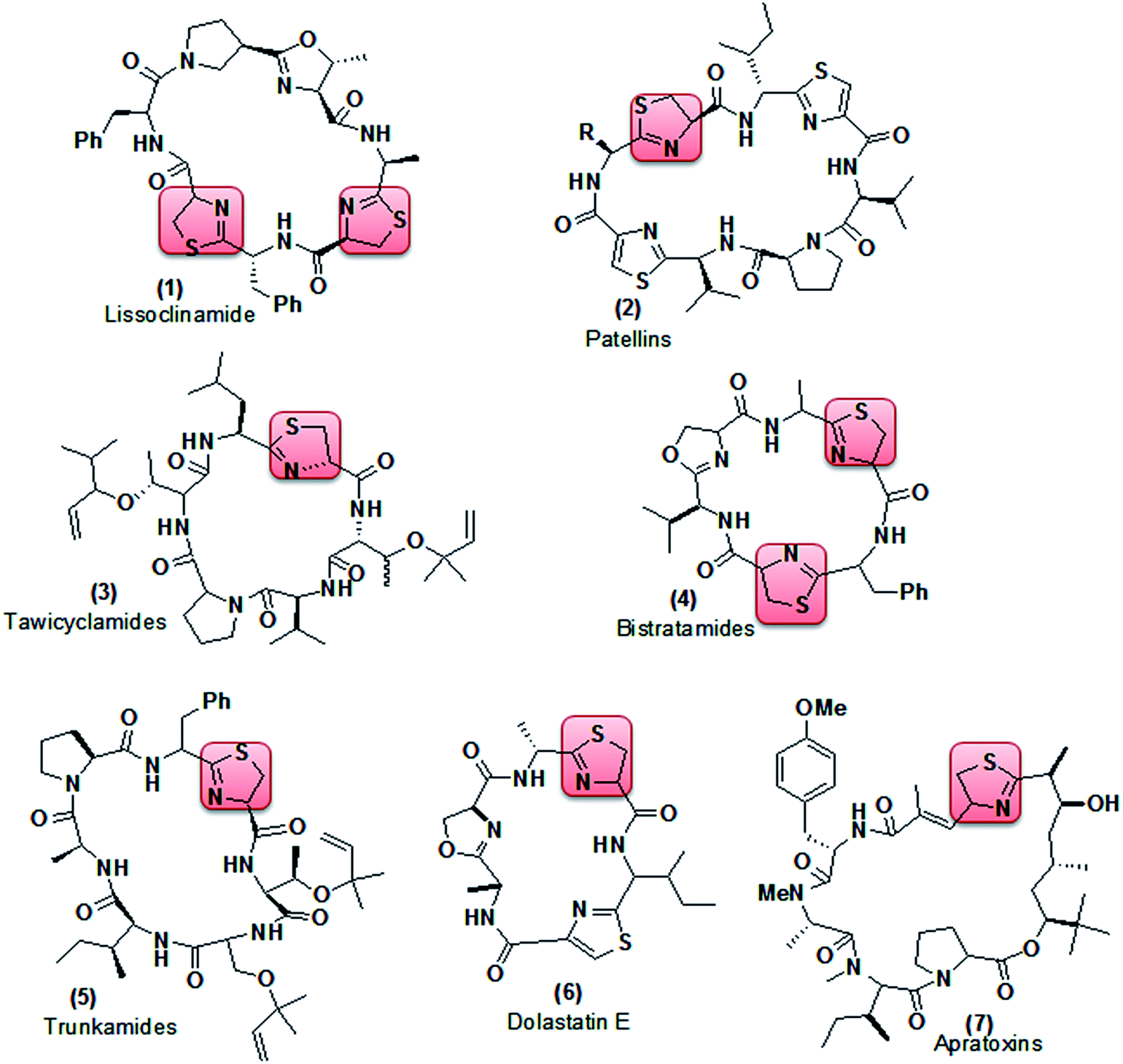
Thiazoline (partially reduced thiazole) is a heterocyclic compound containing both sulfur and nitrogen in the ring. It is a structural segment of various cyclopeptide alkaloids extracted from different marine organisms.7 Lissoclinamide (1, Fig. 2), patellins (2), tawicyclamides (3), bistratamides (4), and trunkamides (5) were extracted from Lissoclinum patella, whereas dolastatin E (6) and apratoxins (7) were isolated from Dolabella auricularia and Lyngbya majuscula8 respectively. Reports have shown that lissoclinamide (1) having two thiazoline rings was found to be the most cytotoxic when tested with human fibroblasts, bladder carcinoma cell lines, and normal lymphocytes (IC50 < 0.1 μg ml−1),9 whereas trunkamide (5) was reported to have promising antitumor activity10 (Fig. 2).
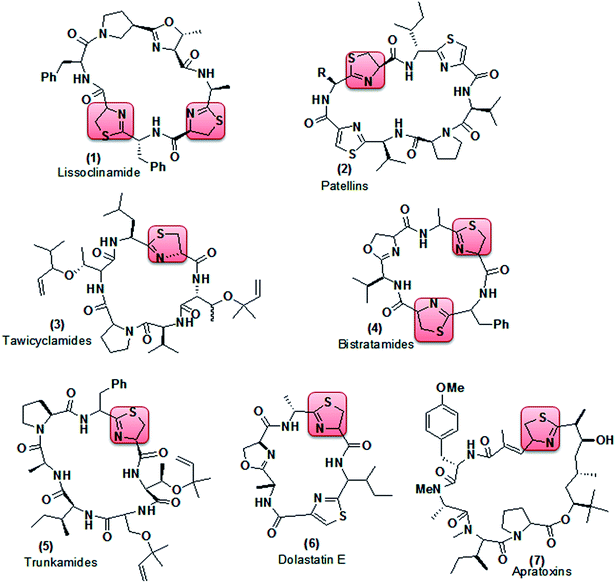 |
| | Fig. 2 Cyclopeptide alkaloids containing thiazoline as a structural segment. | |
Recently, many research groups have paid attention to the chemical properties of thiazolines, mainly due to the unique properties of the sulfur and nitrogen. The derivatives are often bioactive, exhibiting interesting biological activities like antimicrobial, anti-inflammatory, anticancer and anti-HIV activity, pheromone activity and cell division inhibition,11–16 while small molecular mass thiazolines have found applications in the food and flavor industries.17 However, it’s surprising that thiazolines have not been much explored in the field of tubercular activity though they have been found to be good antibiotics.18 Hence, looking into the biological significance of thiazoline particularly in the field of antibiotics, it was desirable for us to introduce thiazoline into the sub-substructure of the compounds by conjugating different coumarin derivatives with thiazoline; since the conjugation of coumarin derivatives with various bioactive molecules such as resveratrol, tacrine, thiazol-pyrazole, triazole, chalcone and pyrimidine has produced novel hybrid molecules, which are endowed with vasorelaxant, platelet anti-aggregating, Alzheimer’s disease treatment, antimicrobial, antioxidant, Hsp90 C-terminal inhibitor, antioxidant, trypanocidal and anti-cancer properties.19 Furthermore, hybridized compounds have diverse or dual modes of action, multiple biological activities, modified selectivity profiles and reduced undesired side effects. Such molecules may be further modified to exhibit favorable pharmacokinetics and oral bioavailability.20
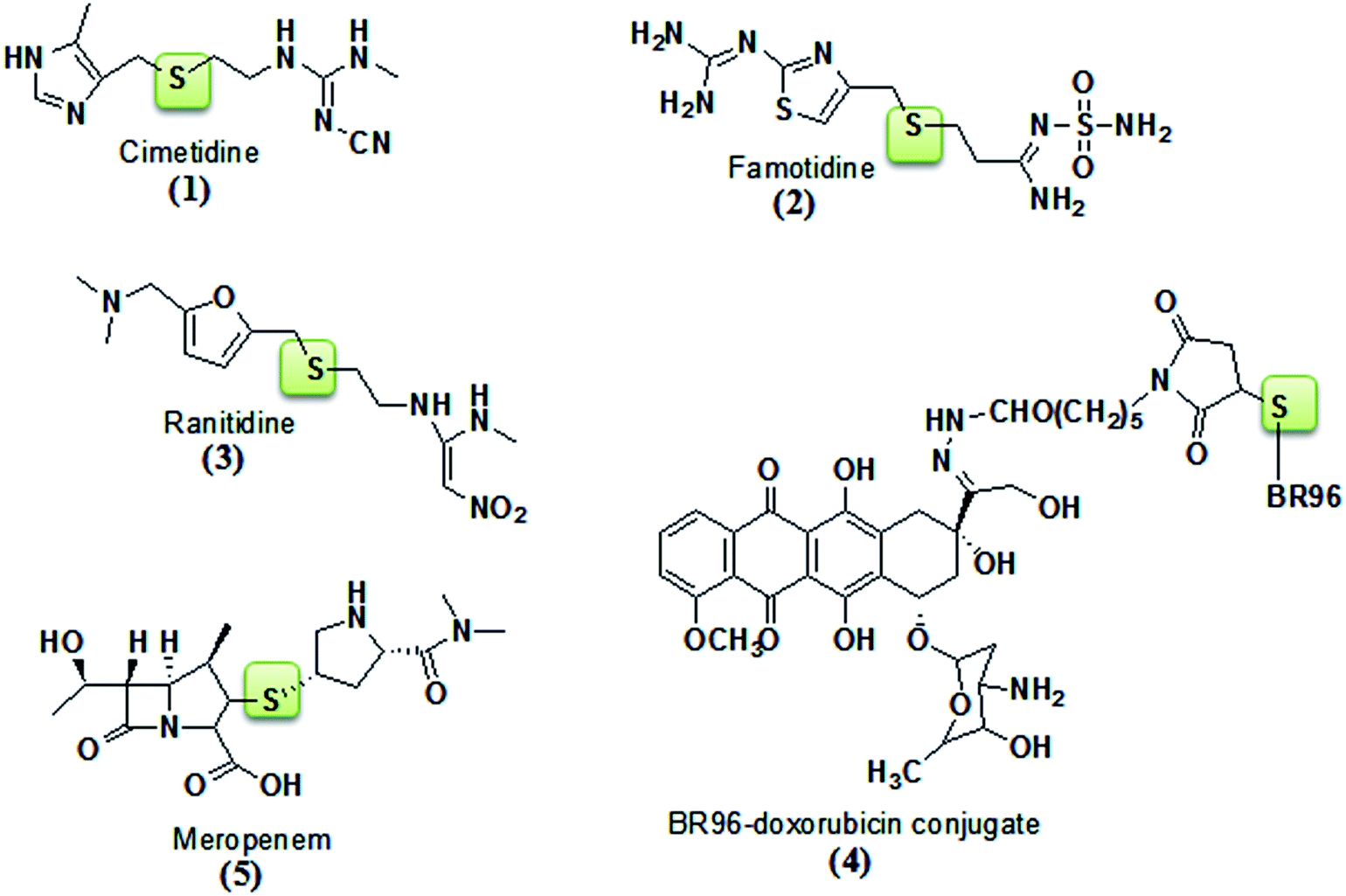
Thioether linkages are present in many bioactive natural and pharmaceutical products. However it’s surprising that in comparison to the amount of effort devoted to the discovery of new methods for preparing C–C, C–O, and C–N bonds, considerably less resources have been allocated to the development of preparing C–S bonds.21 In fact, it has been demonstrated in several instances that replacing a carbon or oxygen atom with sulfur greatly enhances the bioactivity of certain compounds with respect to their oxygenated or carbon counterparts.22 For example, SAR studies have shown that for diallyl sulfide, which exhibits potent anticancer properties,22 a single sulfur atom bonded to at least one allyl side chain is required for inhibition of carcinogenesis.23 A number of drugs, which are applied for the treatment of Alzheimer’s disease, Parkinson’s disease, and diabetes, as well as immune and inflammatory diseases, carry aryl sulfide moieties on their backbone unit.24 Furthermore, the thioether linkage has provided compounds with diverse antagonistic properties against the histamine H2-receptor, e.g. cimetidine25 (Tagamet) (1), famotidine26 (Pepcid) (2), and ranitidine27 (Zantac) (3) are being used to treat and prevent ulcers; the thioether linkage in an immunity-related conjugate between doxorubicin and the monoclonal antibody BR96 was demonstrated to be critical to its antitumor properties28 (4); and meropenem29 (5) has been used as an antibiotic drug (Fig. 3). Hence, it is apparent that the development of novel thioethers, and more generally, C–S bonds, would be of great benefit to both chemists and biologists for developing potent drugs.
 |
| | Fig. 3 Bioactive thioether linked compounds. | |
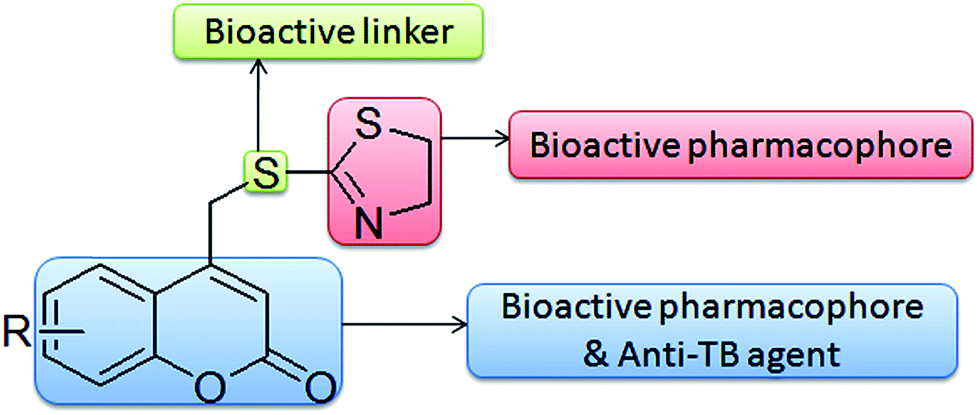
Hence, motivated by the inherent biological relevance of coumarin, thiazoline, and thioether, the present investigation pertains to the hybridization of two active pharmacophores (coumarin & thiazoline) via a thioether linkage. It is expected that the additive effect of this combination might produce a synergetic effect in enhancing the bio-activity of the compound. The designed hypothetical interaction model is represented in (Fig. 4). To further our continued effort towards the development of microwave assisted synthetic methodologies,19f,30 we describe in this paper an efficient and facile synthesis of coumarin–thiazoline hybrids (1a–1j) under microwave irradiation, through which the yields of the compounds were improved drastically in a very short reaction time as compared to conventional methods.
 |
| | Fig. 4 Design strategy to enhance anti-tubercular activity (hypothetical interaction model). | |
Results and discussion
Chemistry
The substituted 4-bromomethyl coumarins (a–j) were synthesized using a Pechman cyclisation of the phenols with 4-bromoethylacetoacetate.31 Condensation of the 4-bromomethyl coumarin (a–j) (0.01 mol) with 4,5-dihydrothiazole-2-thiol (1) (0.01 mol) in anhydrous K2CO3 (0.03 mol) using absolute ethanol as the solvent afforded 4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]substituted-2H-chromen-2-one derivatives (1a–1j) under both conventional and microwave irradiation methods. Synthesis of the target compounds was carried out as outlined in Scheme 1. From the results, it is clear that the microwave approach proved to be extremely fast, providing good to excellent yields (81–91%) as compared to the conventional method (61–75%). Here the most noticeable advancement was the speed with which the reactions were completed i.e. within 5–9 minutes, which is 90–120 times faster than the conventional method. The results are summarized in Table 1.
 |
| | Scheme 1 Synthetic route to synthesize the title compounds (1a–1j). | |
Table 1 Comparison between the conventional and microwave irradiation methods
| Product |
R |
Yield (%) |
Time (min) |
| Ca |
Mb |
C |
M |
| C – conventional. M – microwave. |
| 1a |
6-CH3 |
63 |
88 |
600 |
5 |
| 1b |
6-Cl |
71 |
83 |
630 |
7 |
| 1c |
6-OCH3 |
75 |
91 |
670 |
6 |
| 1d |
5,6-Benzo |
64 |
82 |
810 |
8 |
| 1e |
7-CH3 |
66 |
86 |
640 |
6 |
| 1f |
7-Cl |
68 |
84 |
650 |
7 |
| 1g |
7-OCH3 |
69 |
87 |
720 |
6 |
| 1h |
5,7-diCH3 |
61 |
81 |
840 |
9 |
| 1i |
6-Br |
64 |
88 |
730 |
8 |
| 1j |
7-Br |
62 |
87 |
720 |
8 |
All the newly synthesized compounds were characterized using FTIR, 1H NMR, 13C NMR, mass and elemental analysis. The spectral data of the newly synthesized compounds (1a–1j) are provided in the experimental section, and are in accordance with the assigned structures of the compounds. The 1H and 13C NMR spectra of all the compounds are given in the ESI‡ and are in good agreement with the proposed structure of the compounds. In the case of compound 1a (R = 6CH3), the IR spectrum exhibited two characteristic bands at 1713 cm−1 for the lactone of the coumarin and 1574 cm−1 for the imine of the thiazoline moiety. The formation of the product was established using the 1H NMR spectrum, wherein a sharp singlet at δ 4.45 ppm corresponds to the –CH2–S, confirming the formation of the condensed product via thioether linkage, and the presence of two triplets at δ 3.45 ppm and δ 4.23 ppm corresponds to the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–CH2 and –S–CH2 of the thiazoline ring respectively. Two singlets were observed in the aromatic region at δ 6.56 and δ 7.40 ppm, corresponding to the C3–H and C5–H of the coumarin moiety respectively, whereas the C7–H and C8–H of the coumarin resonate as doublets at δ 7.24 and δ 7.34 ppm respectively, while the methyl protons were observed as a singlet at δ 2.42 ppm which corresponds to the C6–CH3 of the coumarin. The 13C NMR spectrum provides additional support for the structure of the compound (1a), wherein the lactone carbonyl resonated at δ 160.73 ppm, the thioether linked –S–CH2 resonated at δ 36.10 ppm, and the –CN of the imine resonated at δ 163.59 ppm, whereas the –S–CH2 and N–CH2 corresponding to the thiazoline ring resonated at δ 32.06 ppm and δ 63.89 ppm respectively, and the methyl carbon resonated at δ 21.03 ppm which corresponds to the C6–CH3 of the coumarin. The molecular ion peak at 291[M]+ in the GC-MS spectrum confirmed the proposed structure for compound 1a. The rest of the compounds gave satisfactory analytical and spectroscopic data which were in accordance with their assigned structures.
N–CH2 and –S–CH2 of the thiazoline ring respectively. Two singlets were observed in the aromatic region at δ 6.56 and δ 7.40 ppm, corresponding to the C3–H and C5–H of the coumarin moiety respectively, whereas the C7–H and C8–H of the coumarin resonate as doublets at δ 7.24 and δ 7.34 ppm respectively, while the methyl protons were observed as a singlet at δ 2.42 ppm which corresponds to the C6–CH3 of the coumarin. The 13C NMR spectrum provides additional support for the structure of the compound (1a), wherein the lactone carbonyl resonated at δ 160.73 ppm, the thioether linked –S–CH2 resonated at δ 36.10 ppm, and the –CN of the imine resonated at δ 163.59 ppm, whereas the –S–CH2 and N–CH2 corresponding to the thiazoline ring resonated at δ 32.06 ppm and δ 63.89 ppm respectively, and the methyl carbon resonated at δ 21.03 ppm which corresponds to the C6–CH3 of the coumarin. The molecular ion peak at 291[M]+ in the GC-MS spectrum confirmed the proposed structure for compound 1a. The rest of the compounds gave satisfactory analytical and spectroscopic data which were in accordance with their assigned structures.
X-ray diffraction analysis
Single crystals for compounds 1a, 1b, 1e and 1h were developed by slow evaporation of chloroform at room temperature. Compounds 1a, 1b and 1e crystallized under a triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and compound 1h under a monoclinic system with the space group P21/c. The unit cell dimensions for compound 1a are as follows: a = 7.93370(10) Å, b = 8.09230(10) Å, c = 10.7299(2) Å, α = 99.1900(10)°, β = 92.5250(10)°, γ = 96.4970(10)°, Z = 2. For compound 1b the unit cell dimensions are: a = 7.3443(3) Å, b = 7.7814(3) Å, c = 12.6804(4) Å, α = 85.486(2)°, β = 82.226(2)°, γ = 65.984(2)°, Z = 2. For compound 1e the unit cell dimensions are: a = 7.8774(2) Å, b = 8.4986(3) Å, c = 11.2330(3) Å, α = 91.435(2)°, β = 107.376(2)°, γ = 107.927(2)°, Z = 2; whereas for compound 1h the unit cell dimensions were found to be: a = 7.2356(8) Å, b = 7.9426(10) Å, c = 25.027(3) Å, α = 90°, β = 96.742(8)°, γ = 90°, Z = 4. The structures were solved and refined using SHELXS-97.‡32
and compound 1h under a monoclinic system with the space group P21/c. The unit cell dimensions for compound 1a are as follows: a = 7.93370(10) Å, b = 8.09230(10) Å, c = 10.7299(2) Å, α = 99.1900(10)°, β = 92.5250(10)°, γ = 96.4970(10)°, Z = 2. For compound 1b the unit cell dimensions are: a = 7.3443(3) Å, b = 7.7814(3) Å, c = 12.6804(4) Å, α = 85.486(2)°, β = 82.226(2)°, γ = 65.984(2)°, Z = 2. For compound 1e the unit cell dimensions are: a = 7.8774(2) Å, b = 8.4986(3) Å, c = 11.2330(3) Å, α = 91.435(2)°, β = 107.376(2)°, γ = 107.927(2)°, Z = 2; whereas for compound 1h the unit cell dimensions were found to be: a = 7.2356(8) Å, b = 7.9426(10) Å, c = 25.027(3) Å, α = 90°, β = 96.742(8)°, γ = 90°, Z = 4. The structures were solved and refined using SHELXS-97.‡32
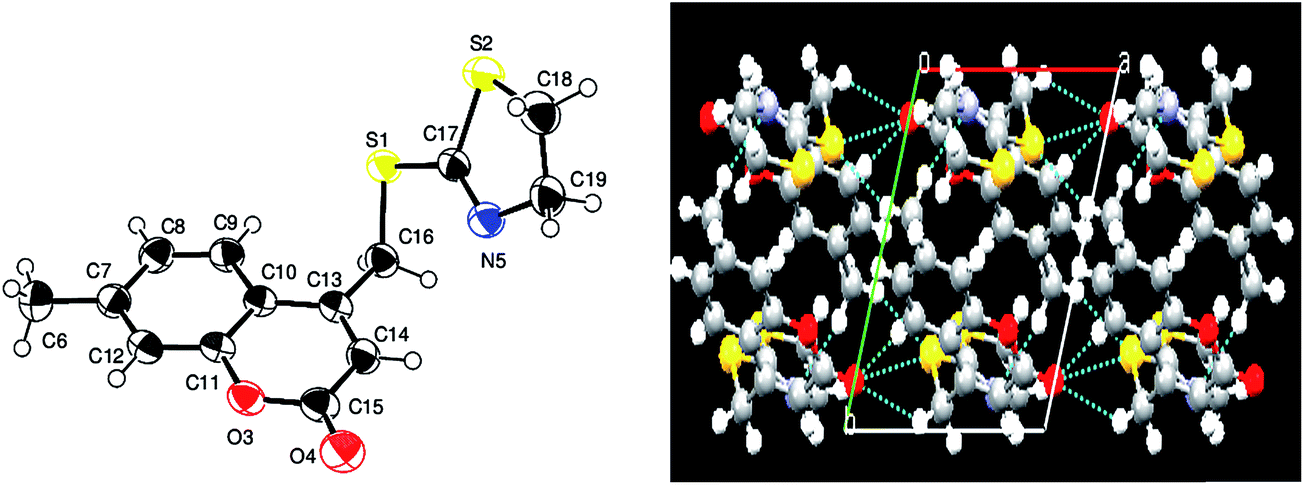
X-ray analysis for compound 1a. From the crystal data it was observed that the asymmetric unit of compound 4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]-6-methyl-2H-chromen-2-one (1a) contains only one independent molecule. The crystal structure shows weak intramolecular C16⋯H16B⋯N5 & C7⋯H7⋯S2 hydrogen bonds and is further stabilized by intermolecular C15–H15C⋯S2 hydrogen bonds, that generate inversion dimers with R22(16) ring motifs. The 2H-chromene ring systems is nearly planar, with a maximum deviation of 0.0234(17) Å for atom C9. In the crystal structure, the bond length C17–S1 = 1.7705(2) Å is longer than the bond length of C18–C19 = 1.498(3) Å and the bond angle C17–S1–C19 = 88.60(10)° is less than the bond angle of C17–N5–C18 = 110.45(9)°. As a result of these differences in the molecular parameters, the thiazoline ring adopts a nearly twisted form conformation. The dihedral angle between the 2H-chromene ring (O3/C6–C14) and the thiazoline ring (N5/S1/C17–C19) is 82.72(10)°. The packing of the crystal structure is stabilized by C19–H19A⋯π Cg(3) (C9–C14) interactions as well as π–π [Cg(2) (O3/C6–C10)⋯Cg(3) (C9–C14)] interactions between the fused benzene and the pyrone ring of the coumarin moieties [shortest centroid–centroid distance = 3.6368 (10) Å]. The packing of the molecules exhibited layered stacking when viewed down the a-axis. These layers are linked via hydrogen bonds, which in turn form a linear polymeric chain. The X-ray structure parameters and refinement for compound (1a) are presented in Table 2. The ORTEP and packing diagrams of compound 1a are portrayed in Fig. 5.
Table 2 Crystal data, data collection details and structure refinement for 1a
| Empirical formula |
C14H13NO2S2 |
| Formula weight |
291.37 |
| Crystal system, space group |
Triclinic, P |
| Unit cell dimensions |
a = 7.93370(10) Å |
| b = 8.09230(10) Å |
| c = 10.7299(2) Å |
| α = 99.1900(10)°, β = 92.5250(10)°, γ = 96.4970(10)° |
| Volume |
674.313(2) Å3 |
| Z |
2 |
| Calculated density |
1.435 mg m−3 |
| Crystal size |
0.22 × 0.15 × 0.12 mm |
| Absorption coefficient |
0.391 mm−1 |
| F(000) |
304 |
| Crystal form |
Plate, colourless |
| Radiation source |
Fine-focus sealed tube |
| Radiation type |
Mo Kα |
| Radiation monochromator |
Graphite |
| Criterion for observed reflection |
I > 2σ(I) |
|
| Data collection |
| Diffractometer |
Bruker SMART CCD area-detector |
| Data collection method |
ω–χ scans |
| Absorption correction |
Multi-scan |
| Theta range for data collection |
1.93 to 24.99° |
| Limiting indices |
−9 ≤ h ≤ 9, −9 ≤ k ≤ 9, −12 ≤ l ≤ 12 |
| Reflections collected/unique |
10734/2376 [R(int) = 0.0200] |
| Completeness to theta |
99.6% |
| Max. and min. transmission |
Tmax = 1.000, Tmin = 0.790 |
|
| Refinement |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
2376/0/172 |
| Goodness-of-fit on F2 |
1.059 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0379, wR2 = 0.1108 |
| R indices (all data) |
R1 = 0.0414, wR2 = 0.1141 |
| Weighting scheme |
ω = 1/[σ2(Fo2) + (0.068P)2 + 0.230P] where P = (Fo2 + 2Fc2)/3 |
| (Δ/σ)max |
<0.001 |
| Largest diff. peak and hole |
0.356 and −0.237e Ǻ−3 |
 |
| | Fig. 5 ORTEP and packing diagrams for compound 1a. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radius. | |
X-ray analysis for compound 1b. From the crystal data it is known that the asymmetric unit of 6-chloro-4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]-2H-chromen-2-one (1b) contains only one independent molecule. The crystal structure shows weak intramolecular C8⋯H8⋯S3 and C16⋯ H16B⋯N6 and intermolecular C16⋯H16A⋯O5 hydrogen bonds and is further stabilized by intermolecular C16–H16B⋯Cl1 hydrogen bonds, that generate inversion dimers with R22(14) ring motifs. The 2H-chromene ring (O4/C7–C15) system is nearly planar, with a maximum deviation of 0.0202(23) Å for atom C13. In the crystal structure, the bond length C17–S2 = 1.7595(2) Å is longer than the bond length of C18–C19 = 1.495(4) Å and the bond angle C17–S2–C19 = 89.31(12)° is less than the bond angle of C17–N6–C18 = 111.0(2)°. As a result of these differences in the molecular parameters, the thiazoline ring adopts a nearly twisted form conformation. The dihedral angle between the 2H-chromene ring (O4/C7–C15) and the thiazoline (S2/N6/C17–C19) ring is 89.16(11)°. The packing of the crystal structure is stabilized by π–π [Cg(2) (O4/C7–C11)⋯Cg(3) (C10–C15)] interactions between the fused benzene and the pyrone ring of the coumarin moieties [shortest centroid–centroid distance = 3.7101 (13) Å]. The crystal structure parameters for compound 1b are presented in Table 3. The ORTEP and packing diagrams of compound 1b are portrayed in Fig. 6.
Table 3 Crystal data, data collection details and structure refinement for 1b
| Empirical formula |
C13H10ClNO2S2 |
| Formula weight |
311.79 |
| Temperature |
296 K |
| Wavelength |
0.71073 Å |
| Crystal system, space group |
Triclinic, P |
| Unit cell dimensions |
a = 7.3443(3) Å |
| b = 7.7814(3) Å |
| c = 12.6804(4) Å |
| α = 85.486(2)°, β = 82.226(2)°, γ = 65.984(2)° |
| Volume |
655.63(4) Å3 |
| Z |
2 |
| Calculated density |
1.579 mg m−3 |
| Crystal size |
0.22 × 0.15 × 0.12 mm |
| Absorption coefficient |
0.61 mm−1 |
| F(000) |
320 |
| Crystal form |
Plate, yellow |
| Radiation source |
Fine-focus sealed tube |
| Radiation type |
Mo Kα |
| Radiation monochromator |
Graphite |
| Criterion for observed reflection |
I > 2σ(I) |
|
| Data collection |
| Diffractometer |
Bruker SMART CCD area-detector |
| Data collection method |
ω–χ scans |
| Absorption correction |
Multi-scan |
| Theta range for data collection |
1.62 to 24.99° |
| Limiting indices |
−8 ≤ h ≤ 8, −9 ≤ k ≤ 9, −15 ≤ l ≤ 15 |
| Reflections collected/unique |
9894/2307 [R(int) = 0.0252] |
| Completeness to theta |
99.8% |
| Max. and min. transmission |
Tmax = 1.000, Tmin = 0.790 |
|
| Refinement |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
2307/0/172 |
| Goodness-of-fit on F2 |
1.075 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0340, wR2 = 0.0921 |
| R indices (all data) |
R1 = 0.0380, wR2 = 0.0946 |
| Weighting scheme |
ω = 1/[σ2(Fo2) + (0.0505P)2 + 0.2154P] where P = (Fo2 + 2Fc2)/3 |
| (Δ/σ)max |
<0.001 |
| Largest diff. peak and hole |
0.338 and −0.319e Ǻ−3 |
 |
| | Fig. 6 ORTEP and packing diagrams for compound 1b. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radius. | |
X-ray analysis for compound 1e. From the crystal data it is known that the asymmetric unit of 4-((4,5-dihydrothiazol-2-ylthio)methyl)-7-methyl-H-chromen-2-one (1e) contains only one independent molecule. The crystal structure shows weak intramolecular C16⋯H16B⋯N5 and C7⋯H7⋯S2 hydrogen bonds. In addition, intermolecular C6⋯H6C⋯N5 hydrogen bonds link the components into chains along [100] and generate inversion dimers with R22(22) ring motifs. The 2H-chromene ring (O3/C7–C15) system is nearly planar, with a maximum deviation of 0.0271(18) Å for atom C14. In the crystal structure, the bond length C18–S2 = 1.802(2) Å is longer than the bond length of C18–C19 = 1.522(3) Å and the bond angle C17–S2–C18 = 88.63(9)° is less than the bond angle of C17–N5–C19 = 111.16(15)°. As a result of these differences in the molecular parameters, the thiazoline ring adopts a nearly twisted form conformation. The dihedral angle between the 2H-chromene ring (O3/C7–C15) and the thiazoline ring (S2/N5/C17–C19) is 63.61(8)°. The crystal structure also features C⋯H⋯π [Cg(2); (O3/C10/C11/C13–C15)] and π–π [Cg(2) (O3/C10/C11/C13–C15)⋯Cg(3) (C7–C12)] interactions between the fused benzene and the pyrone ring of the coumarin moieties [shortest centroid–centroid distance = 3.7698(9) Å] and shows stacking when viewed along the b-axis. The crystal structure parameters for compound 1e are presented in Table 4. The ORTEP and packing diagrams of compound 1e are portrayed in Fig. 7.
Table 4 Crystal data, data collection and structure refinement (1e)
| Empirical formula |
C14H13NO2S2 |
| Formula weight |
291.37 |
| Temperature |
293 K |
| Wavelength |
0.71073 Å |
| Crystal system, space group |
Triclinic, P |
| Unit cell dimensions |
a = 7.8774(2) Å |
| b = 8.4986(3) Å |
| c = 11.2330(3) Å |
| α = 91.435(2)°, β = 107.376(2)°, γ = 107.927(2)° |
| Volume |
677.16(3) Å3 |
| Z |
2 |
| Calculated density |
1.429 mg m−3 |
| Crystal size |
0.22 × 0.15 × 0.12 mm |
| Absorption coefficient |
0.391 mm−1 |
| F(000) |
304 |
| Crystal form |
Plate, colourless |
| Radiation source |
Fine-focus sealed tube |
| Radiation type |
Mo Kα |
| Radiation monochromator |
Graphite |
| Criterion for observed reflection |
I > 2σ(I) |
|
| Data collection |
| Diffractometer |
Bruker SMART CCD area-detector |
| Data collection method |
ω–χ scans |
| Absorption correction |
Multi-scan |
| Theta range for data collection |
1.92 to 29.02° |
| Limiting indices |
−10 ≤ h ≤ 10, −11 ≤ k ≤ 11, −15 ≤ l ≤ 15 |
| Reflections collected/unique |
13042/3587 [R(int) = 0.0236] |
| Completeness to theta |
99.4% |
| Max. and min. transmission |
Tmax = 1.000, Tmin = 0.790 |
|
| Refinement |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3587/0/172 |
| Goodness-of-fit on F2 |
1.04 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0419, wR2 = 0.1165 |
| R indices (all data) |
R1 = 0.0526, wR2 = 0.1247 |
| Weighting scheme |
ω = 1/[σ2(Fo2) + (0.0653P)2 + 0.1591P] where P = (Fo2 + 2Fc2)/3 |
| (Δ/σ)max |
<0.001 |
| Largest diff. peak and hole |
0.508 and −0.490e Ǻ−3 |
 |
| | Fig. 7 ORTEP and packing diagrams for compound 1e. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radius. | |
X-ray analysis for compound 1h. From the crystal data it is known that the asymmetric unit for compound 4-((4,5-dihydrothiazol-2-ylthio)methyl)-5,7-dimethyl-2H-chromen-2-one (1h) is characterized by a long range, well defined three dimensional order. The asymmetric unit contains only one independent molecule as depicted in Fig. 8. The 2H-chromene ring (O3/C11/C12/C14–C16) system is nearly planar, with a maximum deviation of 0.0162(13) Å for atom C13, and in the crystal structure, the bond length C20–S2 = 1.802(6) Å is longer than the bond length of C19–C20 = 1.509(8) Å and the bond angle C18–S2–C20 = 88.0(3)° is less than the bond angle of C18–N5–C19 = 109.6(5)°. As a result of these differences in the molecular parameters, the thiazoline ring adopts a nearly twisted form conformation. The dihedral angle between the 2H-chromene ring and the thiazoline ring (S2/N5/C18–C20) is 8.1(3)°. The crystal structure contains weak intramolecular C15⋯H15⋯S1 hydrogen bonds. In addition, intermolecular C13⋯H13⋯O4 hydrogen bonds link the components into chains along [010] and generate inversion dimers with R22(12) ring motifs. The crystal structure also features C7⋯H7B⋯π [Cg(3); (C8–C13)] and π–π [Cg(2) (O3/C11/C12/C14–C16)⋯Cg(3) (C8–C13)] interactions between the fused benzene and the pyrone ring of the coumarin moieties [shortest centroid–centroid distance = 3.568(3) Å] and shows stacking when viewed along the c-axis. The crystal structure parameters for compound 1h are presented in Table 5. The ORTEP and packing diagrams of compound 1h are portrayed in Fig. 8.
 |
| | Fig. 8 ORTEP and packing diagrams for compound 1h. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms are shown as spheres of arbitrary radius. | |
Table 5 Crystal data, data collection details and structure refinement for 1h
| Empirical formula |
C15H15NO2S2 |
| Formula weight |
305.40 |
| Temperature |
296 K |
| Wavelength |
0.71073 Å |
| Crystal system, space group |
Monoclinic, P21/c |
| Unit cell dimensions |
a = 7.2356(8) Å |
| b = 7.9426(10) Å |
| c = 25.027(3) Å |
| α = 90°, β = 96.742(8)°, γ = 90° |
| Volume |
1428.4(3) Å3 |
| Z |
4 |
| Calculated density |
1.420 mg m−3 |
| Crystal size |
0.24 × 0.20 × 0.12 mm |
| Absorption coefficient |
0.373 mm−1 |
| F(000) |
640 |
| Crystal form |
Plate, yellow |
| Radiation source |
Fine-focus sealed tube |
| Radiation type |
Mo Kα |
| Radiation monochromator |
Graphite |
| Criterion for observed reflection |
I > 2σ(I) |
|
| Data collection |
| Diffractometer |
Bruker SMART CCD area-detector |
| Data collection method |
ω–χ scans |
| Absorption correction |
Multi-scan |
| Theta range for data collection |
1.64 to 22.29° |
| Limiting indices |
−7 ≤ h ≤ 7, −8 ≤ k ≤ 8, −26 ≤ l ≤ 26 |
| Reflections collected/unique |
6121/1743 [R(int) = 0.0536] |
| Completeness to theta |
95.2% |
| Max. and min. transmission |
Tmax = 1.000, Tmin = 0.770 |
|
| Refinement |
| Refinement method |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
1743/0/181 |
| Goodness-of-fit on F2 |
1.071 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0710, wR2 = 0.1526 |
| R indices (all data) |
R1 = 0.0954, wR2 = 0.1644 |
| Weighting scheme |
ω = 1/[σ2(Fo2) + (0.039P)2 + 4.4284P] where P = (Fo2 + 2Fc2)/3 |
| (Δ/σ)max |
<0.001 |
| Largest diff. peak and hole |
0.309 and −0.357e Ǻ−3 |
Bioevaluation
The synthesized compounds were screened for their potential in vitro anti-tubercular activity using a Microplate Alamar Blue Assay (MABA)33 and in a CT-DNA cleavage study using a gel electrophoresis method.34 Furthermore, the most active compounds were tested for their cytotoxicity against Vero cells using a MTT35 assay.
Anti-tubercular activity. All the compounds (1a–1j) were initially screened at a single concentration of 6.25 μg ml−1 against MtbH37Rv (ATCC-27294) in BACTEC 12 B medium, using a Microplate Alamar Blue Assay (MABA). The results are summarized in Table 6. Compounds exhibiting ≥90% inhibition in the initial screening were tested at and below 6.25 μg ml−1 using 2-fold dilution to determine the actual MIC. The results of the anti-tubercular studies are presented in Table 6. In the primary screening, most of the compounds displayed 91–98% inhibition. In the secondary level of screening, four compounds (1b, 1f, 1i and 1j) inhibited Mtb with a MIC < 1 μg ml−1 and two compounds (1a and 1c) had a MIC < 4 μg ml−1, compared to isoniazid (MIC: 0.02 μg ml−1). From Table 6, it is clear that the halogen (–Cl and –Br) substituted compounds (1b, 1f, 1i and 1j) have shown more significant anti-tubercular activity, with MICs in the range 0.09–0.78 μg ml−1. Among them, compounds 1b and 1i exhibited the most pronounced anti-tubercular activity with a MIC of 0.09 and 0.19 μg ml−1 respectively. In particular, compound 1b was found to be the most active in vitro with a MIC of 0.09 μg ml−1. Whereas compounds 1a and 1c exhibited comparatively good activity with a MIC 1.56 and 3.12 μg ml−1 respectively, while compounds 1e and 1g showed moderate inhibition against Mtb with similar MIC values of 6.25 μg ml−1. The remaining compounds 1d and 1h were found to be ineffective against the MtbH37Rv strain. Although from Table 6, no clear relation was observed between the MIC and the lipophilicity (calculated logP).
Table 6 In vitro anti-tubercular screening data of the compounds (1a–1j) against MtbH37Rv
| Compound |
R |
% inhibition at a concentration of 6.25 μg ml−1 |
MICa (μg ml−1) |
% survival of Vero cells at a concentration of 10 × MICb |
logPc |
| Minimum inhibitory concentration against the H37Rv strain of Mtb (μg ml−1). A compound is considered toxic if it causes over 50% inhibition of normal cells at a concentration 10 fold higher than its MIC. Calculated using http://www.molinspiration.com/; N.D – not determined. |
| 1a |
6-CH3 |
93 |
1.56 |
76% |
3.16 |
| 1b |
6-Cl |
98 |
0.09 |
94% |
3.38 |
| 1c |
6-OCH3 |
92 |
3.12 |
72% |
2.76 |
| 1d |
5,6-Benzo |
16 |
N.D |
N.D |
3.89 |
| 1e |
7-CH3 |
91 |
6.25 |
64% |
3.16 |
| 1f |
7-Cl |
94 |
0.39 |
88% |
3.38 |
| 1g |
7-OCH3 |
92 |
6.25 |
61% |
2.76 |
| 1h |
5,7-diCH3 |
23 |
N.D |
N.D |
3.53 |
| 1i |
6-Br |
96 |
0.19 |
91% |
3.52 |
| 1j |
7-Br |
94 |
0.78 |
81% |
3.52 |
| Isoniazid |
— |
100 |
0.02 |
98% |
−0.97 |
Having identified a good number of active antimycobacterial compounds (1a, 1b, 1c, 1e, 1f, 1g, 1i and 1j) the next step was to examine their toxicity against the Vero cell line, at a concentration 10 times their actual MIC value. A compound is considered toxic if it causes over 50% inhibition of normal cells at a concentration 10 fold higher than its MIC.36 From the results (Table 6), it is clear that the tested compounds exhibited moderate to low levels of cytotoxicity with a percentage survival of the Vero cells in the range of 61 to 94%, and none of the tested compounds exhibited any significant cytotoxic effects on the Vero cells, suggesting great potential for their in vivo use as anti-tubercular agents. As for the activity against Vero cells, the highest cytotoxicity was caused by having –CH3 (1e) and –OCH3 (1g) substituents at the C-7 position of the coumarin, with a percentage survival of 64 and 61% respectively. A second level of cytotoxicity was observed for the –CH3 (1a) and –OCH3 (1c) substituents at the C-6 position, with a percentage survival of 76 and 72% respectively. Whereas, the halogen-substituted compounds (1b, 1f, 1i and 1j) exhibited the highest safety profiles with a percentage survival of the Vero cells in the range of 81–94%, which suggest that these compounds can act as new leads for the development of new anti-tubercular drugs. Fig. 9 gives a comparison of the % survival of the Vero cells at a concentration of the compound which is 10 fold higher than its actual MIC value (μg ml−1).
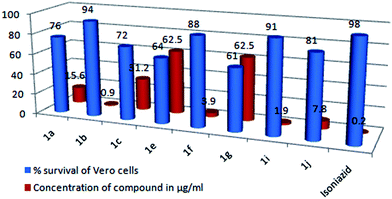 |
| | Fig. 9 Comparison of the percentage survival of the Vero cells at a concentration of the compound which is 10 times its actual MIC value (μg ml−1). | |
Preliminary SAR study
Even though the number of compounds tested here is limited, a few key features regarding the structural requirements for the coumarin–thiazoline hybrids (1a–1j) to exert their anti-tubercular activity may be determined. As we have discussed in the Introduction section, our initial strategy was to identify the key sub-units required for activity such as the coumarin, thiazoline and thioether linker. Additionally, the substituents, like R = –CH3 (electron donating), –OCH3 (electron releasing), –Br and –Cl (halogen), were varied at the C-6 and C-7 positions of the coumarin, since modification of these positions is more favorable for activity and the best output results were obtained with substituents at these particular sites.5c,37 A fused benzo substituent at the 5,6 position of the coumarin (1d) was also involved in the series, since benzo groups fused to coumarins have been reported in photocleavage studies,38 and their electronic along with their photo-physical characteristics have resulted in promising biological applications.39 Hence based on the structural data the following assumptions about the structure–activity relationship (SAR) were outlined.
From the results, it is clear that the group of compounds having halogen (–Cl & –Br) substituents (1b, 1f, 1i and 1j) have shown more significant anti-tubercular activity with MICs in the range 0.09–0.78 μg ml−1. Among them the halogen (–Cl & –Br) substituents at the C-6 position of the coumarin moiety (1b and 1i) greatly enhanced the activity of the compound, obtaining MIC values of 0.09 and 0.19 μg ml−1 respectively; in particular, the –Cl substituent at the C-6 position was found to be the most active in vitro with a MIC of 0.09 μg ml−1. A slight change in the position of the halogen from the C-6 to C-7 position decreases the overall activity of the compound, obtaining MIC values of 0.39 and 0.78 μg ml−1 for 1f and 1j respectively. A second line of activity was shown by the –CH3 (1a) and –OCH3 (1b) substituents at the C-6 position of the coumarin obtaining MIC values of 1.56 and 3.12 μg ml−1 respectively, whereas those compounds with –CH3 (1e) and –OCH3 (1g) at the C-7 position of the coumarin exhibited moderate activity with a similar MIC value of 6.25 μg ml−1. Here the substituents at the C-6 position have shown much better results than those at the C-7 position of the coumarin moiety; while the 5,6-benzo (1d) and 5,7-diCH3 (1h) substituents were found to be inactive against the MtbH37Rv strain. From the above results it is evident that the halogen substituents dominated the overall activity. The reason for this increase in activity may be attributed to the high lipophilic nature of the halogen and it being less water soluble, hence penetration of the drug or drug lead into a cell wall of lipid membrane is much more easy and thereby the bioavailability of the compound is increased.40 However, the other substituents (–CH3, –OCH3, 5,6-benzo & 5,7-CH3) have shown mixed activity and no proper SAR could be explained for these substituents. Although it is worth mentioning that the –CH3, –OCH3 and halogen substituents (–Cl & –Br) at the C-6 position were found to be more active than the same substituents at the C-7 position of the coumarin ring. The results from the preliminary structure–activity analysis have led to the determination of some key structural requirements for the coumarin–thiazoline hybrids to exert their anti-TB activity, which provides insight for further structural modifications.
Electrophoresis analysis
The design and development of small- or medium-sized potential therapeutic agents to target nucleic acid cleavage can lead to the synthesis of novel therapeutic agents for infectious diseases and can act as a tool for molecular biology. Interaction of small molecules with specific sites along a DNA strand as a reactive model for protein–nucleic acid bindings provides a path toward rational drug design as well as a means to develop sensitive chemical probes for DNA. The different loci present in DNA are involved in various regulatory processes such as gene expression, gene transcription, mutagenesis, carcinogenesis, etc.41 In particular, designing of the compound based on its ability to cleave DNA is of great importance not only from the primary biological point of view but also in terms of a photodynamic therapeutic approach to develop potent drugs.42 Hence to understand the mode of action of the synthesized compounds (1a–1j) and to identify new molecules that induce apoptosis, i.e. programmed cell death, we carried out a CT-DNA cleavage assay using an agarose gel electrophoresis method, and the results are presented in Fig. 10. The cleavage potential of the test compounds was assessed by comparing the bands that appeared for the control and test compounds at 5 μM concentration. The DNA alone without the test compound was used as a control, which did not show any cleavage of DNA even after a long exposure time. From the photograph (Fig. 10) it is clear that compounds 1b, 1d, 1f and 1i cleaved the DNA more efficiently, as no traces of DNA were found suggesting that the cleavage of DNA by the compounds may be attributed to the denaturation of double-stranded DNA by cleavage of the hydrogen bonds between the nitrogen bases and the phosphodiester bonds between the pentose sugars at various sites. In addition, cleavage of the DNA might have occurred frequently leading to low molecular weight fragments and these small fragments have migrated away from the gel during electrophoresis. The remaining compounds 1a, 1c, 1g, 1h and 1j were found to be inactive for cleavage of DNA, whereas a little tailing of the band can be observed in the sample treated with compound 1e indicating shearing of the DNA. From a structural point of view it is observed that the halogen substituents at the 6 and 7 position of the coumarin moiety (1b, 1f, 1i and 1j) exhibited nearly similar MIC values with distinct antitubercular activity in the range 0.09–0.78 μg ml−1, whereas in the DNA cleavage assay, except for the –Br substituent at the 7 position (1j), all the remaining halogen substituent compounds (1b, 1f and 1i) cleave the DNA more efficiently. This indicates that the nuclease activity may be dependent on the position of the –Br group. Also, if we compare the MIC values of all the halogen-substituted compounds, the compound with a –Br substituent at the 7 position (1j) exhibited the least activity with a MIC of 0.78 μg ml−1. Hence, this slight decrease in activity of compound 1j may be attributed to not cleaving the genomic DNA of the MtbH37 strain or that the target site of compound 1j may be any other cell organelles of the MtbH37 strain but not DNA. It is also noted that compound 1d, which was inactive against the MtbH37 strain, cleaved DNA more efficiently. This indicated that the nuclease activity is dependent on the substituent attached to the coumarin moiety. Furthermore, the concentration of compound 1d used for the in vitro assay may be insufficient to reach the target site, since in the DNA cleavage assay the genomic DNA is directly subjected to the tested compound whereas the MIC study is related to the resistance of the strain (MtbH37) to the tested compound due to many factors. However, the nature of the reactive intermediates involved in the DNA cleavage by the compounds has not been clear.
 |
| | Fig. 10 CT-DNA cleavage study using agarose gel electrophoresis. Lane M: standard DNA marker, Lane C: control DNA (untreated sample), Lane 1a: DNA + 1a, Lane 1b: DNA + 1b, Lane 1c: DNA + 1c, Lane 1d: DNA + 1d, Lane 1e: DNA + 1e, Lane 1f: DNA + 1f, Lane 1g: DNA + 1g, Lane 1h: DNA + 1h, Lane 1i: DNA + 1i, Lane 1j: DNA + 1j. | |
Materials and methods
Instrumentation
Melting points were determined using an open capillary method on a Buchi apparatus and are uncorrected. IR spectra were recorded on a Nicolet 5700 FT-IR instrument (Nicolet, Madison, WI, USA) using KBr discs. 1H NMR spectra were recorded on a Bruker 400 MHz spectrometer using CDCl3 as the solvent and TMS as an internal standard. All chemical shifts are reported as δ values (ppm). Mass spectra were recorded using a Shimadzu GCMSQP2010S. The elemental analyses were carried out using a Hereaus CHN rapid analyzer. The microwave irradiation syntheses were carried out using a CEM-Discover Focused Microwave system. The purity of the compounds was checked using TLC. The X-ray single-crystal structures of compounds 1a, 1b, 1e and 1h were recorded using a Bruker Smart CCD and refined using the SHELXL Software Package.
General procedure for the preparation of compounds 1a–1j
Conventional method. A mixture of 4,5-dihydrothiazole-2-thiol (0.01 mol) and powdered anhydrous K2CO3 (0.03 mol) with the substituted 4-bromomethyl coumarin (0.01 mol) in 5 ml absolute ethanol was stirred at room temperature for 10–14 h. The progress of the reaction was examined using thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was quenched in crushed ice; the solid product was filtered and washed with water. Lastly, the product was recrystallized from chloroform.
Microwave method. A mixture of 4,5-dihydrothiazole-2-thiol (0.01 mol) and powdered anhydrous K2CO3 (0.03 mol) with the substituted 4-bromomethyl coumarin (0.01 mol) was put into a 10 ml microwave pressure vial and irradiated in a microwave oven (model: CEM-Discover Focused Microwave system) under 100 W power at 55 °C for 5–9 min in 5 ml absolute ethanol. The progress of the reaction was examined using thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was quenched in crushed ice; the solid product was filtered and washed with water. Lastly the product was recrystallized from chloroform.
Characterization of the compounds
4-[(4,5-Dihydro-1,3-thiazol-2-ylthio)methyl]-6-methyl-2H-chromen-2-one (1a). Colourless crystals; mp 165–167 °C; IR (KBr) (vmax/cm−1): 1713 (CO of lactone), 1574 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 2.42 (s, 3H, CH3), 3.45 (t, 2H, CH2, J = 8 Hz), 4.23 (t, 2H, CH2, J = 8 Hz), 4.45 (s, 2H, CH2), 6.56 (s, 1H, CH), 7.24 (d, 1H, Ar-H, J = 8.4 Hz), 7.34 (dd, 1H, Ar-H, J = 8.6 Hz, 2 Hz), 7.40 (s, 1H, Ar-H); 13C NMR (100 MHz, CDCl3, δ ppm): 21.03, 32.06, 36.10, 63.89, 115.95, 117.10, 117.91, 124.01, 132.96, 134.00, 149.99, 151.96, 160.73, 163.59; GC-MS: 291[M]+; anal. calcd for C14H13NO2S2: C, 57.71; H, 4.50; N, 4.81%. Found: C, 57.68; H, 4.54; N, 4.85%.
6-Chloro-4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]-2H-chromen-2-one (1b). Yellow crystals; mp 181–183 °C; IR (KBr) (vmax/cm−1): 1716 (CO of lactone), 1577 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.55 (t, 2H, CH2, J = 8 Hz), 4.33 (t, 2H, CH2, J = 8 Hz), 4.55 (s, 2H, CH2), 6.96 (s, 1H, CH), 7.31 (d, 1H, Ar-H, J = 8.4 Hz), 7.54 (d, 1H, Ar-H, J = 8.4 Hz), 7.66 (s, 1H, Ar-H); 13C NMR (100 MHz, CDCl3, δ ppm): 31.95, 36.05, 64.11, 116.13, 117.43, 118.21, 123.96, 133.06, 133.92, 150.08, 152.53, 160.11, 162.94; GC-MS: 311[M]+, 313[M + 2]+; anal. calcd for C13H10ClNO2S2: C, 50.08; H, 3.23; N, 4.49%. Found: C, 50.05; H, 3.28; N, 4.54%.
4-((4,5-Dihydrothiazol-2-ylthio)methyl)-6-methoxy-2H-chromen-2-one (1c). Colourless crystals; mp 163–165 °C; IR (KBr) (vmax/cm−1): 1713 (CO of lactone), 1584 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.37 (t, 2H, CH2, J = 8 Hz), 3.73 (s, 3H, –OCH3), 4.15 (t, 2H, CH2, J = 8 Hz), 4.52 (s, 2H, CH2), 6.48 (s, 1H, CH), 7.14 (d, 1H, Ar-H, J = 8.4 Hz), 7.28 (dd, 1H, Ar-H, J = 8.4 Hz, 1.6 Hz), 7.41 (s, 1H, Ar-H); 13C NMR (100 MHz, CDCl3, δ ppm): 32.40, 37.70, 55.44, 62.15, 115.40, 117.51, 118.97, 124.00, 132.01, 133.75, 149.91, 152.06, 159.73, 162.97; GC-MS: 307[M]+; anal. calcd for C14H13NO3S2: C, 54.70; H, 4.26; N, 4.56%. Found: C, 54.75; H, 4.30; N, 4.60%.
1-((4,5-Dihydrothiazol-2-ylthio)methyl)-3H-benzo[f]chromen-3-one (1d). Brown solid; mp 190–192 °C; IR (KBr) (vmax/cm−1): 1707 (CO of lactone), 1601 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.43 (t, 2H, CH2, J = 8 Hz), 4.22 (t, 2H, CH2, J = 8 Hz), 4.49 (s, 2H, CH2), 6.61 (s, 1H, CH), 7.53–7.66 (m, 4H), 7.82 (dd, 1H, J = 8.4 Hz, 1.6 Hz), 8.48 (d, 1H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3, δ ppm): 32.58, 36.10, 63.91, 113.55, 115.20, 119.76, 122.56, 123.21, 124.30, 127.22, 127.63, 128.80, 134.75, 151.01, 151.07, 160.60, 163.53; GC-MS: 327[M]+; anal. calcd for C17H13NO2S2: C, 62.36; H, 4.00; N, 4.28%. Found: C, 62.39; H, 4.04; N, 4.25%.
4-((4,5-Dihydrothiazol-2-ylthio)methyl)-7-methyl-2H-chromen-2-one (1e). Colourless crystals; mp 167–169 °C; IR (KBr) (vmax/cm−1): 1713 (CO of lactone), 1597 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 2.38 (s, 3H, CH3), 3.41 (t, 2H, CH2, J = 8 Hz), 4.19 (t, 2H, CH2, J = 8 Hz), 4.41 (s, 2H, CH2), 6.51 (s, 1H, –CH), 7.18 (d, 1H, J = 8.4 Hz), 7.30 (dd, 1H, J = 8.4 Hz, 1.6 Hz), 7.37 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 21.01, 32.05, 36.11, 63.88, 115.90, 117.05, 117.90, 124.02, 132.95, 134.01, 150.02, 151.92, 160.67, 163.55; GC-MS: 291[M]+; anal. calcd for C14H13NO2S2: C, 57.71; H, 4.50; N, 4.81%. Found: C, 57.75; H, 4.54; N, 4.83%.
7-Chloro-4-((4,5-dihydrothiazol-2-ylthio)methyl)-2H-chromen-2-one (1f). Yellow crystals; mp 183–185 °C; IR (KBr) (vmax/cm−1): 1711 (CO of lactone), 1579 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.45 (t, 2H, CH2, J = 8 Hz), 4.22 (t, 2H, CH2, J = 8 Hz), 4.41 (s, 2H, CH2), 6.58 (s, 1H), 7.27 (d, 1H, J = 8.8 Hz), 7.46 (dd, 1H, J = 8.6 Hz, 2 Hz), 7.64 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 31.89, 36.23, 63.76, 117.01, 118.73, 119.36, 124.05, 129.79, 132.01, 149.34, 152.25, 159.80, 163.44; GC-MS: 311[M]+, 313[M + 2]+; anal. calcd for C13H10ClNO2S2: C, 50.08; H, 3.23; N, 4.49%. Found: C, 50.05; H, 3.27; N, 4.45%.
4-((4,5-Dihydrothiazol-2-ylthio)methyl)-7-methoxy-2H-chromen-2-one (1g). Colourless crystals; mp 165–167 °C; IR (KBr) (vmax/cm−1): 1716 (CO of lactone), 1589 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.40 (t, 2H, –CH2, J = 8 Hz), 3.83 (s, 3H, –OCH3), 4.21 (t, 2H, CH2, J = 8 Hz), 4.41 (s, 2H, CH2), 6.59 (s, 1H), 7.20 (d, 1H, Ar-H, J = 8 Hz), 7.31 (s, 1H, J = 8.4 Hz), 7.40 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 32.75, 36.66, 56.75, 63.08, 116.18, 117.14, 118.81, 123.75, 134.01, 134.68, 148.74, 152.31, 161.03, 163.42; GC-MS: 307[M]+; anal. calcd for C14H13NO3S2: C, 54.70; H, 4.26; N, 4.56%. Found: C, 54.75; H, 4.24; N, 4.60%.
4-((4,5-Dihydrothiazol-2-ylthio)methyl)-5,7-dimethyl-2H-chromen-2-one (1h). Yellow crystals; mp 182–184 °C; IR (KBr) (vmax/cm−1): 1709 (CO of lactone), 1597 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 2.29 (s, 3H, CH3), 2.64 (s, 3H, CH3), 3.41 (t, 2H, CH2, J = 8 Hz), 4.16 (t, 2H, CH2, J = 8 Hz), 4.42 (s, 2H, CH2), 6.46 (s, 1H), 6.82 (s, 1H), 6.98 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 21.14, 23.95, 35.99, 36.15, 63.86, 115.25, 116.51, 116.57, 130.17, 135.57, 142.19, 151.29, 155.73, 160.42, 163.78; GC-MS: 305[M]+; anal. calcd for C15H15NO2S2: C, 58.99; H, 4.95; N, 4.59%. Found: C, 58.75; H, 4.89; N, 4.63%.
6-Bromo-4-((4,5-dihydrothiazol-2-ylthio)methyl)-2H-chromen-2-one (1i). Yellow crystals; mp 175–177 °C; IR (KBr) (vmax/cm−1): 1715 (CO of lactone), 1601 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.51 (t, 2H, CH2, J = 8 Hz), 4.14 (t, 2H, CH2, J = 8 Hz), 4.56 (s, 2H, CH2), 6.61 (s, 1H), 7.34 (d, 1H, J = 8 Hz), 7.41 (d, 1H, J = 7.6 Hz),7.60 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 32.11, 36.01, 64.13, 117.12, 117.46, 120.16, 127.15, 133.63, 137.26, 149.81, 153.75, 161.13, 163.78; GC-MS: 311[M]+, 313[M + 2]+; anal. calcd for C13H10BrNO2S2: C, 43.83; H, 2.83; N, 3.93%. Found: C, 43.89; H, 2.87; N, 3.98%.
7-Bromo-4-((4,5-dihydrothiazol-2-ylthio)methyl)-2H-chromen-2-one (1j). Yellow crystals; mp 179–181 °C; IR (KBr) (vmax/cm−1): 1718 (CO of lactone), 1583 (CN of thiazole) cm−1; 1H NMR (400 MHz, CDCl3, δ ppm) δ 3.43 (t, 2H, CH2, J = 8 Hz), 4.21 (t, 2H, CH2, J = 8 Hz), 4.48 (s, 2H, CH2), 6.61 (s, 1H), 7.41 (d, 1H, J = 8.4 Hz), 7.61 (dd, 1H, J = 7.6 Hz, 1.6 Hz), 7.82 (s, 1H); 13C NMR (100 MHz, CDCl3, δ ppm): 33.09, 36.13, 64.22, 116.22, 117.49, 119.13, 126.82, 131.18, 135.12, 150.09, 153.49, 161.46, 164.03; GC-MS: 311[M]+, 313[M + 2]+; anal. calcd for C13H10BrNO2S2: C, 43.83; H, 2.83; N, 3.93%. Found: C, 43.80; H, 2.89; N, 3.97%.
Anti-tubercular assay
All the compounds were first screened at a concentration of 6.25 μg ml−1 against M. tuberculosis H37Rv (ATCC-27294) in BACTEC 12 B medium using the Microplate Alamar Blue Assay (MABA). Compounds exhibiting <90% inhibition in the primary screening were not evaluated further, while compounds exhibiting >90% inhibition were re-tested against MtbH37Rv at lower concentrations in order to determine the actual minimum inhibitory concentration (MIC) in the MABA. The experiments were carried out in triplicate using a 96-well plate; to each well 100 μl of Middlebrook 7H9 broth was added and a serial dilution of the compound was made directly on the plate. Firstly, 200 ml of sterile deionized water was added to all outer perimeter wells of a sterile 96-well plate to minimize evaporation of the medium in the test wells during incubation. Isoniazid was included as a positive drug control. The test compounds were serially diluted using a two-fold serial dilution method. The plates were covered and sealed with para film and incubated at 37 °C for 5 days. After this, 25 ml of a freshly prepared 1:1 mixture of Alamar Blue reagent and 10% tween 80 was added to the plate and incubated for 24 h. A blue color in the wells indicated inhibition of bacterial growth while a pink color was recorded as growth. Furthermore, the minimum concentration of the compound required to inhibit the bacterial growth was determined.
Assay for in vitro cytotoxicity against Vero cells
The cytotoxicity of the compounds with a MIC ≤ 6.25 μg ml−1 was evaluated using Vero cells. The Vero cells were cultured in Dulbecco Modified Eagle Medium (DMEM) containing 2 mM Na2CO3 supplemented with 10% (v/v) fetal bovine serum (FBS). The cells were incubated at 37 °C under 5% CO2 and 95% air in a humidified atmosphere until confluent and then diluted with phosphate-buffered saline to obtain 106 cells per ml. Stock solutions were prepared in dimethyl sulfoxide (DMSO) and further dilutions were made with fresh culture medium. The medium was removed and replaced by 180 ml of fresh medium containing the test compound at a concentration 10 fold its actual MIC value. After incubation at 37 °C for 72 h, the medium was removed and the monolayer was washed twice with 100 μL of warm Hanks’ balanced salt solution (HBSS). One hundred microliters of warm medium and 20 μL of freshly made MTS-PMS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium and phenylmethasulfazone] (100:20) (Promega) were added to each well, the plates were incubated for 3 h, and the absorbance was determined at 560 nm using a microplate reader. The percentage of cell survival was calculated after considering the control wells (cells incubated in DMSO-containing medium).
Sample preparation
Calf-Thymus DNA (CT-DNA) was procured from Merck Bangalore. A DNA stock solution was prepared by diluting the CT-DNA in TE buffer. The test compounds (5 μM) were dissolved in DMSO and added separately to the 10 μl of CT-DNA and then incubated at 37 °C for 2 h.
Agarose gel electrophoresis
The cleavage of Calf-Thymus DNA (CT-DNA) by the products was analyzed using an agarose gel electrophoresis method. The gel was prepared by adding 200 mg of agarose in 25 ml of TAE buffer (4.84 g Tris base, pH 8.0, 0.5 M EDTA per 1 liter). The mixture was heated to dissolve the agarose completely; thereafter it was slightly cooled and 5 μl of ethidium bromide was added. Then the molten agarose was poured into a casting tray and a comb was placed in it for the formation of wells and it was allowed to solidify. After solidification the comb was removed and the tray was placed in a tank wherein the tank buffer (TAE) was poured. Then the wells were loaded with 10 μl of DNA, 5 μm of the test compound and 10 μl of the tracking dye (10 mM tris pH 8.0, 1 mM EDTA, 30% glycerol, 0.2% bromophenol blue). Electrophoresis was performed at 50 volts for 45 min. The gel was visualized on a UV transilluminator for analysis of the cleavage of the treated DNA sample by using untreated DNA as a control and a marker was used to examine the molecular weight. All the experiments were carried out in triplicate under the same conditions.
Conclusion
The present study describes the synthesis of 4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]substituted-2H-chromen-2-one derivatives (1a–1j) under microwave irradiation which gives excellent yields in a shorter reaction time as compared to the conventional method. The most noticeable advancement in the present study is the speed at which the reaction was completed i.e. 5–9 minutes, which was 90–120 times faster than the conventional method. Single X-ray crystals of compounds 1a, 1b, 1e and 1h were developed and their crystal parameters were evaluated, which can contribute to the understanding of the reactivity, affinity and binding properties of the molecules. Anti-tubercular activity screening revealed that compound 6-chloro-4-[(4,5-dihydro-1,3-thiazol-2-ylthio)methyl]-2H-chromen-2-one (1b) was found to be the most active in vitro, with a MIC of 0.09 μg ml−1, and it exhibited a low level of cytotoxicity against Vero cells, which suggests that compound 1b can act as promising lead for the development of new anti-tubercular agents. Furthermore, compounds 1b and 1i which were found to be the most active in vitro against the MtbH37 strain were found to cleave DNA completely, which further confirmed that compounds 1b and 1i are biologically more potent in vitro agents. The obtained results suggest that the potent compounds may serve as lead chemical entities for further modification in the search for new classes of potential anti-tubercular drugs.
Acknowledgements
This research work is financially supported by the UGC, New Delhi under the UPE-FAR-I program, F. no. 14-3/2012 (NS/PE) and the Council of Scientific & Industrial Research (CSIR), New Delhi – 110 012 under reference no. 5496/NS/EMR-II/2015. The authors thank USIC, Karnatak University Dharwad, for providing spectral and X-ray analysis. We are also grateful to the NMR Research Centre, Indian Institute of Science (IISc), Bengaluru, India for the recording of the 1H and 13C NMR spectra.
References
-
(a) J. D. Bos and M. M. H. M. Meinardi, Exp. Dermatol., 2000, 9, 165–169 CAS;
(b) D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
-
(a) C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed;
(b) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed.
-
(a) X. M. Peng, G. L. V. Damu and C. H. Zhou, Curr. Pharm. Des., 2013, 19, 3884–3930 CrossRef CAS PubMed;
(b) S. H. Bairagi, P. P. Salaskar, S. D. Loke, N. N. Surve, D. V. Tandel and M. D. Dusara, Int. J. Pharm. Res., 2012, 4, 16–19 CAS;
(c) C. Kontogiorgis, A. Detsi and D. Hadjipavlou-Litina, Expert Opin. Ther. Pat., 2012, 22, 437–454 CrossRef CAS PubMed;
(d) S. Mirunalini and M. Krishnaveni, Int. J. PharmTech Res., 2011, 3, 1693–1696 CAS.
-
(a) S. A. Stanley, T. Kawate, N. Iwase, M. Shimizu, A. Clatworthy, E. Kazyanskaya, J. C. Sacchettini, T. R. Ioerger, N. Siddiqi, S. Minami, J. A. Aquadro, S. S. Grant, E. J. Rubin and D. T. Hung, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 11565–11570 CrossRef CAS PubMed;
(b) M. Jeyachandran, P. Ramesh, D. Sriram, P. Senthilkumar and P. Yogeeswari, Bioorg. Med. Chem. Lett., 2012, 22, 4807–4809 CrossRef CAS PubMed;
(c) M. Basanagouda, V. B. Jambagi, N. N. Barigidad, S. S. Laxmeshwar and V. Devaru Narayanachar, Eur. J. Med. Chem., 2014, 74, 225–233 CrossRef CAS PubMed;
(d) I. Ahmad, J. P. Thakur, D. Chanda, D. Saikia, F. Khan, S. Dixit, A. Kumar, R. Konwar, A. S. Negi and A. Gupta, Bioorg. Med. Chem. Lett., 2013, 23, 1322–1325 CrossRef CAS PubMed.
-
(a) T. Kawate, N. Iwase, M. Shimizu, S. A. Stanley, S. Wellington, E. Kazyanskaya and D. T. Hung, Bioorg. Med. Chem. Lett., 2013, 23, 6052–6059 CrossRef CAS PubMed;
(b) J. S. Mossa, E. Feraly and I. Muhammad, Phytother. Res., 2004, 18, 934–937 CrossRef CAS PubMed;
(c) G. Appendino, E. Mercalli, N. Fuzzati, L. Arnoldi, M. Stavri, S. Gibbons, M. Ballero and A. Maxia, J. Nat. Prod., 2004, 67, 2108–2110 CrossRef CAS PubMed;
(d) M. Figueroa, R. I. Cruz, R. B. Cruz, R. Byeb, A. Navarrete and R. Mata, J. Ethnopharmacol., 2007, 113, 125–131 CrossRef CAS PubMed;
(e) C. C. Chiang, M. J. Cheng, C. F. Peng, H. Y. Huang and I. S. Chen, Chem. Biodiversity, 2010, 7, 1728–1736 CrossRef CAS PubMed.
-
(a) P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS;
(b) B. S. Davidson, Chem. Rev., 1993, 93, 1771–1791 CrossRef CAS.
-
(a) H. Luesch, W. A. Yoshida, R. E. Moore, V. J. Paul and T. H. Corbett, J. Am. Chem. Soc., 2001, 123, 5418–5423 CrossRef CAS PubMed;
(b) H. Luesch, W. A. Yoshida, R. E. Moore and V. J. Paul, Bioorg. Med. Chem., 2002, 10, 1973–1978 CrossRef CAS PubMed.
- C. J. Hawkins, M. F. Lavin, K. A. Marshall, A. L. van den Brenk and D. J. Watters, J. Med. Chem., 1990, 33, 1634–1638 CrossRef CAS PubMed.
-
(a) W. Konigsberg, R. J. Hill and L. C. Craig, J. Org. Chem., 1961, 26, 3867–3871 CrossRef CAS;
(b) K. Yonetani, Y. Hirotsu and T. Shiba, Bull. Chem. Soc. Jpn., 1975, 48, 3302–3305 CrossRef CAS.
- A. C. Gaumont, M. Gulea and J. Levillain, Chem. Rev., 2009, 109, 1371–1401 CrossRef CAS PubMed.
- E. Pontiki, D. Hadjipavlou-Litina, A. T. Chaviarab and C. A. Bolosb, Bioorg. Med. Chem. Lett., 2006, 16, 2234–2237 CrossRef CAS PubMed.
- P. Wipf and P. C. Fritch, Tetrahedron Lett., 1994, 35, 5397–5400 CrossRef CAS.
- R. J. Boyce, G. C. Mulqueen and G. Pattende, Tetrahedron Lett., 1994, 35, 5705–5708 CrossRef CAS.
- S. D. Sharrow, M. V. Novotny and M. J. Stone, Biochemistry, 2003, 42, 6302–6309 CrossRef CAS PubMed.
- J. Y. Lai, J. Yu, B. Mekonnen and J. R. Falck, Tetrahedron Lett., 1996, 37, 7167–7170 CrossRef CAS.
-
(a) K. Kumazawa, Food Sci. Technol. Res., 2006, 12, 71–84 CrossRef CAS;
(b) A. Adams and N. De Kimpe, Chem. Rev., 2006, 106, 2299–2313 CrossRef CAS PubMed.
-
(a) H. Fujimoto, T. Kinoshita, H. Suzuki and H. Umezawa, J. Antibiot., 1970, 23, 271–275 CrossRef CAS PubMed;
(b) S. Pestka and N. Brot, J. Biol. Chem., 1971, 246, 7715–7722 CAS;
(c) D. J. W. Burns, Eur. J. Biochem., 1973, 37, 570–574 CrossRef CAS PubMed;
(d) A. Ino and A. Murabayashi, Tetrahedron, 1999, 55, 10271–10282 CrossRef CAS;
(e) L. C. Craig, W. F. Phillips and M. Burachik, Biochemistry, 1969, 8, 2348–2356 CrossRef CAS PubMed;
(f) H. Yamaguchi, Y. Nakayama, K. Takeda, K. Tawara, K. Maeda, T. Takeuchi and H. Umezawa, J. Antibiot., 1957, 10, 195–200 CAS.
-
(a) S. Vilar, E. Quezada, L. Santana, E. Uriarte, M. Yanez, N. Fraiz, C. Alcaide, E. Cano and F. Orallo, Bioorg. Med. Chem. Lett., 2006, 16, 257–261 CrossRef CAS PubMed;
(b) S. S. Xie, X. Wang, N. Jiang, W. Yu, K. D. G. Wang, J. S. Lan, Z. R. Li and L. Y. Kong, Eur. J. Med. Chem., 2015, 95, 153–165 CrossRef CAS PubMed;
(c) R. Gondru, J. Banothu, R. K. Thatipamula, S. K. Althaf Hussain and R. Bavantula, RSC Adv., 2015, 5, 33562–33569 RSC;
(d) J. Zhao, H. Zhao, J. A. Hall, D. Brown, E. Brandes, J. Bazzill, P. T. Grogan, C. Subramanian, G. Vielhauer, M. S. Cohen and B. S. J. Blagg, Med. Chem. Commun., 2014, 5, 1317–1323 RSC;
(e) S. V. Rodriguez, R. F. Guíñez, M. J. Matos, L. Santana, E. Uriarte, M. Lapier, J. D. Maya and C. Olea-Azar, Med. Chem. Commun., 2013, 4, 993–1000 RSC;
(f) K. M. Hosamani, D. S. Reddy and H. C. Devarajegowda, RSC Adv., 2015, 5, 11261–11271 RSC.
- S. Sandhu, Y. Bansal, O. Silakari and G. Bansal, Bioorg. Med. Chem., 2014, 22, 3806–3814 CrossRef CAS PubMed.
- R. Forest and W. Jimmy, Org. Lett., 2010, 12, 2668–2671 CrossRef PubMed.
-
(a) J. Arunakaran, A. Arunkumar, M. Vijayababu, P. Venkataraman and K. Senthilkumar, Biol. Pharm. Bull., 2006, 29, 375–379 CrossRef;
(b) M. J. Wargovich, C. Woods, V. W. Eng, L. C. Stephens and K. Gray, Cancer Res., 1988, 48, 6872–6875 CAS;
(c) M. J. Wargovich and H. Sumiyoshi, Cancer Res., 1990, 50, 5084–5087 Search PubMed;
(d) Y. Shukla, A. Arora and I. Siddiqui, Mol. Cancer Ther., 2004, 3, 1459–1466 Search PubMed;
(e) C. Yang, S. Chhabra, J. Hong and T. Smith, J. Nutr., 2001, 131, 1041S–1045S CAS;
(f) T. Ariga and T. Seki, BioFactors, 2006, 26, 93–103 CrossRef CAS PubMed;
(g) Y. Surh, R. Lee, K. Park, S. Mayne, A. Liem and J. Miller, Carcinogenesis, 1995, 16, 2467–2471 CrossRef CAS PubMed.
- M. J. Wargovich, J. Nutr., 2006, 136, 832S–834S CAS.
-
(a) G. Liu, J. R. Huth, E. T. Olejniczak, R. P. Mendoza, S. DeVries, E. B. Reilly, G. F. Okasinski, E. Nielsen, S. W. Fesik and T. W. Geldern, J. Med. Chem., 2001, 44, 1202–1210 CrossRef CAS PubMed;
(b) S. F. Nielsen, E. O. Nielsen, G. M. Olsen, T. Liljefors and D. Peters, J. Med. Chem., 2000, 43, 2217–2226 CrossRef CAS PubMed.
- R. Brimblecombe, W. Duncan, G. Durant, J. Emmett, C. Ganellin and M. Parsons, J. Int. Med. Res., 1975, 3, 86–92 CAS.
- J. M. Howard, A. N. Chremos, M. J. Collen, K. E. McArthur, J. A. Cherner, P. N. Maton, C. A. Ciarleglio, M. J. Cornelius, J. D. Gardner and R. T. Jensen, Gastroenterology, 1985, 88, 1026–1033 CAS.
- J. Bradshaw, R. T. Brittain, J. W. Clitherow, M. J. Daly, D. Jack, B. J. Price and R. Stables, Br. J. Pharmacol., 1979, 66, 464P CAS.
- P. Trail, D. Willner, S. Lasch, A. Henderson, S. Hofstead, A. Casazza, R. Firestone, I. Hellstrom and K. Hellstrom, Science, 1993, 261, 212–215 CAS.
- K. J. Shin, K. D. Koo, K. H. Yoo, Y. K. Kang, S. W. Park and D. J. Kim, Bioorg. Med. Chem. Lett., 2001, 11, 2397–2399 CrossRef CAS PubMed.
- R. S. Keri, K. M. Hosamani, H. S. Reddy and R. V. Shingalapur, Catal.Lett., 2009, 131, 552–559 CrossRef CAS.
- M. V. Kulkarni, B. J. Pujar and V. D. Patil, Arch. Pharm., 1983, 316, 15–21 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELX97 Program for Crystallography Refinement, University of Göttingen, Germany, 1997 Search PubMed.
- S. G. Franzblau, R. S. Witzig, J. C. McLaughlin, P. Torres, G. Madico, A. Hernandez, M. T. Degnan, M. B. Cook, V. K. Quenzer, R. M. Ferguson and R. H. Gilman, J. Clin. Microbiol., 1998, 36, 362–366 CAS.
- T. A. Brown, Essential Molecular Biology: A Practical Approach, Oxford University Press, 1990, vol. 1, p. 51 Search PubMed.
- K. Falzari, Z. Zhu, D. Pan, H. Liu, P. Hongmanee and S. G. Franzblau, Antimicrob. Agents Chemother., 2005, 49, 1447–1454 CrossRef CAS PubMed.
- K. P. Barot, S. V. Jain, N. Gupta, L. Kremer, S. Singh, V. B. Takale, K. Joshi and M. D. Ghate, Eur. J. Med. Chem., 2014, 83, 245–255 CrossRef CAS PubMed.
-
(a) V. B. Jadhav, S. K. Nayak, T. N. Guru Rowb and M. V. Kulkarni, Eur. J. Med. Chem., 2010, 45, 3575–3580 CrossRef CAS PubMed;
(b) M. Ghate, R. A. Kusanur and M. V. Kulkarni, Eur. J. Med. Chem., 2005, 40, 882–887 CrossRef CAS PubMed;
(c) H. M. Revankar, M. V. Kulkarni, S. D. Joshi and U. A. More, Eur. J. Med. Chem., 2013, 70, 750–757 CrossRef CAS PubMed.
-
(a) M. J. G. Fernandes, M. S. T. Goncalves and S. P. G. Costa, Tetrahedron, 2008, 64, 3032–3038 CrossRef CAS;
(b) M. J. G. Fernandes, M. S. T. Goncalves and S. P. G. Costa, Tetrahedron, 2008, 64, 11175–11179 CrossRef CAS;
(c) A. M. Soares, S. P. Costa and M. S. Goncalves, Amino Acids, 2010, 39, 121–133 CrossRef CAS PubMed;
(d) M. J. G. Fernandes, S. P. G. Costa and M. S. T. Goncalves, Tetrahedron, 2011, 67, 2422–2426 CrossRef CAS.
-
(a) C. Tablet and M. Hillebrand, J. Photochem. Photobiol., A, 2007, 189, 73–79 CrossRef CAS;
(b) J. A. Key, S. Koh, Q. K. Timerghazin, A. Brown and C. W. Cairo, Dyes Pigm., 2009, 82, 196–203 CrossRef CAS.
- G. Thomas, Medicinal Chemistry an Introduction, John Wiley & Sons, West Sussex, UK, 2000 Search PubMed.
- S. Sreelatha, P. R. Padma and M. Umadevi, Food Chem. Toxicol., 2009, 47, 702–708 CrossRef CAS PubMed.
- V. Kumar, K. Kaur, D. N. Karelia, V. Beniwal, G. K. Gupta, A. K. Sharma and A. K. Gupta, Eur. J. Med. Chem., 2014, 81, 267–276 CrossRef CAS PubMed.
Footnotes |
| † This research paper is dedicated to our beloved Prof. M. V. Kulkarni on his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 897299, 897300, 1059231 and 1059232. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra09508e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.