DOI:
10.1039/C5RA09464J
(Paper)
RSC Adv., 2015,
5, 60354-60364
Synthesis and antioxidant activity of DOPA peptidomimetics by a novel IBX mediated aromatic oxidative functionalization†
Received
20th May 2015
, Accepted 7th July 2015
First published on 7th July 2015
Abstract
DOPA peptidomimetics with stable O–C and N–C covalent bonds between amino acid residues have been prepared by aromatic oxidative functionalization of tyrosine with 2-iodoxybenzoic acid (IBX). The reaction involves the Michael-like nucleophilic addition of different oxygen and nitrogen protected amino acids on a reactive DOPA quinone intermediate. Similar results were obtained in heterogeneous conditions using supported IBX-amide for more runs. Among the novel derivatives, compounds containing glycine residues showed a more pronounced antioxidant activity in the 2,2-diphenyl picrylhydrazyl (DPPH) radical scavenging cell free assay. Instead, valine derivatives showed the highest biological effect in L5178Y mouse lymphoma cells, by assessing the ability to reduce H2O2 induced DNA breakage in the alkaline comet assay.
1. Introduction
Peptidomimetics are small molecules which are designed to mimic a naturally occurring peptide. They are typically obtained by modification of parent peptides or by total synthesis1 in order to optimize pharmacological properties, such as bioavailability and biological activity.2 The modifications can involve N-alkylation, Cα-substitution, cyclization, N-replacement, carbonyl replacement, heterocyclic generation, Cα-replacement, and backbone or side-chain transformations, as well as the incorporation of unnatural amino acids.3 Among peptidomimetics, DOPA derivatives play a crucial role in the therapy of Parkinson disease (PD). PD is one of the most important neurodegenerative disorder, characterized by dopamine (DA) depletion in dopaminergic neurons of the striatum of the brain, inducing rigidity, tremor, and postural instability as some of the most important symptoms.4 DOPA peptides are able to increase the capacity of DOPA in penetration of the blood brain barrier (BBB)5 by specific peptide-mediated carrier transport systems (PMCTS), thus restoring adequate DA concentration and inhibiting oxidative cell damage.6 They also act as pro-drugs, preserving DOPA from fast metabolic decarboxylation and avoiding the peripheral DA-related side effects.7 L-DOPA-L-Phe is absorbed more efficiently than L-DOPA via the peptide transporter in Caco-2 cells, which are considered to be a good model for in vivo intestinal absorption in humans.8 In a similar way, D-Phe-L-DOPA showed 31-fold higher oral bioavailability and anti-Parkinson activity than L-DOPA in rats.9 DOPA peptides and peptidomimetics are usually synthesized by solution or solid phase procedures, which show a different degree of complexity depending on the method used for the activation/protection of amino acids.10,11 Irrespective to experimental conditions, these syntheses requires tedious and long time protecting/deprotecting steps and have, in principle, an intrinsic low selectivity. This study is focused on the design of a novel synthetic procedure for the preparation of DOPA peptidomimetics by oxidative side chain modification of amino acid residues.12–17 In this context, DOPA-peptides have been previously synthesized with complete stereochemical integrity by oxidation of Tyr residues with tyrosinase from Agaricus bisporus in organic solvent.18 1-hydroxy-1-oxo-1H-1λ5-benz[d][1,2]iodoxol-3-one (2-iodoxybenzoic acid, IBX) was also used in similar trensformations.19,20 IBX performs the ortho-hydroxylation of phenol to catechols, with a selectivity similar to natural polyphenol oxidases.21–25 The regioselectivity of the oxidation is a consequence of the concerted intramolecular oxygen transfer, from iodine (V) in λ5-iodanyl intermediate (I), to ortho-position of the phenol moiety, with concomitant reduction to λ3-iodanyl orthoquinol monoketal (II) (Scheme 1).26
 |
| | Scheme 1 Mechanism of oxidation of phenol by IBX. | |
In this reaction, the chirality of L-DOPA residues is not affected, the L-enantiomer being the only stereoisomer obtained.27 The replacement of the natural amide bond with more stable covalent linkages In DOPA peptides can significantly improve the bioavailability and activity.28,29 It is well known that Tyr residues cross-link to protein receptors under laccase and tyrosinase oxidation, by nucleophilic addition of sulfhydryl groups.30–34 As a general trend, very complex protein agglomerates are obtained, often used as glues.35 Although few informations are available for the structure of these products, it is expected that the reaction proceeds through nucleophilic addition on reactive DOPA ortho-quinone intermediate following the Michael-like 1-4-regiochemistry.36 With the aim to synthesize new L-DOPA-peptidomimetics characterized by stable O–C and N–C bonds, we report here the IBX mediated aromatic oxidative functionalization of Tyr with different oxygen and nitrogen protected α-amino acids. The procedure is able to oxidize the phenol moiety to catechol group with concomitant introduction of the α-amino acid residues on the aromatic ring, exploiting the reactivity of the DOPA-quinone intermediate.37 The general synthetic pathway is described in Scheme 2.
 |
| | Scheme 2 General reactivity scheme of Tyr with IBX in the presence of protected α-amino acids. Step A: oxidation of Tyr to DOPA-quinone. Step B: in situ Michael-like 1-4-addition of protected α-amino acids on DOPA-quinone intermediate, followed by a reduction step. | |
2. Results and discussions
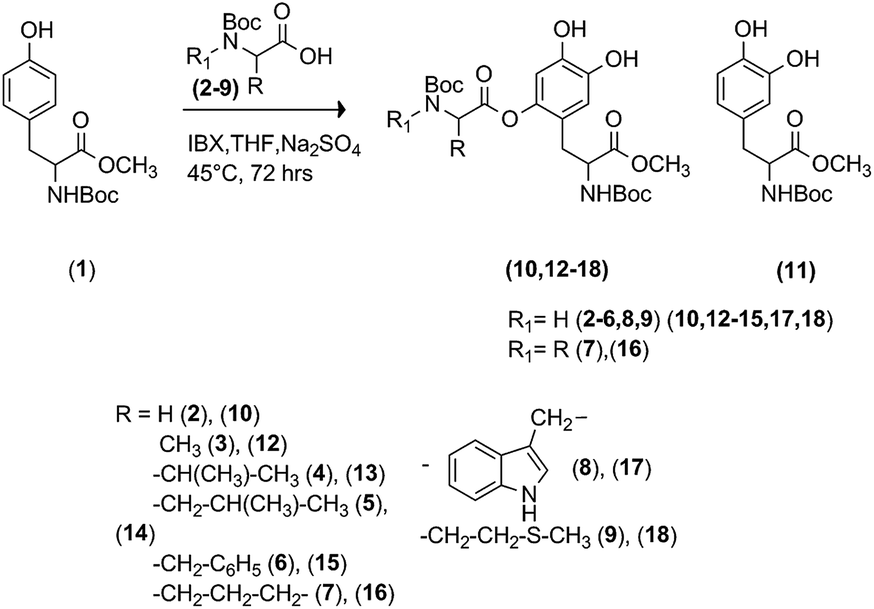
2.1. Synthesis of O–C bonded L-DOPA peptidomimetics
Following the general procedure in Scheme 1, we analyzed the reactivity of N-Boc-Tyr-OMe (1) with a panel of N-Boc protected α-amino acids, including glycine (N-Boc-Gly, 2), alanine (N-Boc-Ala, 3), valine (N-Boc-Val, 4), leucine (N-Boc-Leu, 5), phenylalanine (N-Boc-Phe, 6), proline (N-Boc-Pro, 7), tryptophan (N-Boc-Trp, 8) and methionine (N-Boc-Met, 9). The formation of the L-DOPA quinone intermediate was obtained applying the experimental conditions previously reported for the oxidation of Tyr to L-DOPA with IBX.19 Briefly, compound 1 (0.1 mmol) was dissolved in THF (1.5 mL) in the presence of an excess of (2) (1.0 mmol, as selected nucleophile) and treated with IBX (0.3 mmol) at 25 °C for 3.0 h. The color turned into brown-orange, that is characteristic for the formation of quinone species. After work-up and purification procedures, the desired N-Boc-Gly-N-Boc-DOPA-OMe (10) was obtained in low yield, besides to unreacted substrate and N-Boc-DOPA-OMe (11) (Scheme 2, Table 1, entry 1). In the 1H-NMR of 10, the presence of two aromatic hydrogens (singlets at 7.24 and 7.42 ppm), associated to that of all expected side-chain absorption signals (i.g. two OMe moieties at 3.74 and 4.11, and the Gly CH2 group at 3.93 ppm) confirmed the mono-substitution pattern. Better results were obtained increasing the temperature (45 °C) and the reaction time (72 h) to afford 10 in higher conversion and product yield (Table 1, entry 2). On the basis of these data, the reaction was extended to α-amino acids 3–9 to obtain the corresponding L-DOPA peptidomimetics N-Boc-Ala-N-Boc-DOPA-OMe (12), N-Boc-Val-N-Boc-DOPA-OMe (13), N-Boc-Leu-N-Boc-DOPA-OMe (14), N-Boc-Phe-OMe (1) with N-protected α-amino acids.
Table 1 Synthesis of O–C bonded L-DOPA peptidomimetics
| Entry |
Amino acid |
R |
R1 |
Products |
Conversion (%) |
Yieldsa (%) |
| Reaction conditions: compound 1 (0.1 mmol) was dissolved in THF (1.5 mL) in the presence of the appropriate protected α-amino acids 2–9 (1.0 mmol) and treated with IBX (0.3 mmol) at 45 °C for 72 h. Reaction performed at 25 °C for 3 h. |
| 1 |
2 |
H |
H |
10(11) |
70 |
21(19)b |
| 2 |
2 |
H |
H |
10(11) |
≥98 |
70(10) |
| 3 |
3 |
CH3 |
H |
12(11) |
≥98 |
65(15) |
| 4 |
4 |
CH(CH3)2 |
H |
13(11) |
≥98 |
58(8) |
| 5 |
5 |
CH2CH(CH3)2 |
H |
14(11) |
≥98 |
56(10) |
| 6 |
6 |
CH2C6H5 |
H |
15(11) |
≥98 |
50(16) |
| 7 |
7 |
CH2CH2CH2 |
|
16(11) |
≥98 |
52(12) |
| 8 |
8 |
 |
H |
17(11) |
≥98 |
51(12) |
| 9 |
9 |
CH2CH2SCH3 |
H |
18(11) |
≥98 |
57(3) |
In the case of aliphatic α-amino acids, the yield increased by decreasing of the steric hindrance of the side chain (Table 1, entries 2 and 3 versus entries 4 and 5). Note that, possible side-products due to IBX side-chain oxidation of others low redox potential residues (e.g. tryptophan), were not detected in the reaction mixture. This result is in accordance with the high selectivity of IBX towards the oxidation of phenolic aromatic moieties.38
2.2. Synthesis of N–C bonded L-DOPA peptidomimetics
The procedure was generalized by the use of α-amino acid methyl ester derivatives Gly-OMe (19), Ala-OMe (20), Val-OMe (21), Leu-OMe (22), Phe-OMe (23), Pro-OMe (24), Trp-OMe (25), Met-OMe (26) to synthesize N–C bonded L-DOPA peptidomimetics. The oxidation of compound 1 (0.1 mmol) with IBX (0.3 mmol) in THF (1.5 mL) in the presence of 19 (1.0 mmol) afforded Gly-N-Boc-DOPA-OMe (27) in low yield, besides to traces of N-Boc-DOPA-OMe (11) (Scheme 4, Table 2, entry 1). The reactivity of IBX can be tuned by the use of organic solvents with different properties, the efficacy of the oxidation depending on the nature of the substrate.39 For this reason, we repeated the reaction in the presence of MeOH (1.5 mL) as an alternative solvent, under previously described experimental conditions. As reported in Table 2 (entry 2 versus entry 1), the reaction in MeOH afforded compound 27 in highest yield and conversion of substrate. Further oxidations were then performed with both THF and MeOH solvents. Better results were obtained for aliphatic α-amino acids 20–22 in MeOH, to afford Ala-N-Boc-DOPA-OMe (28), Val-N-Boc-DOPA-OMe (29), and Leu-N-Boc-DOPA-OMe (30) in appreciable yield and quantitative conversion of substrate (Table 2, entries 2, 4, 6 and 8 versus entries 1, 3, 5 and 7).
Table 2 Synthesis of N–C bonded L-DOPA peptidomimeticsa
| Entry |
Amino acid |
Solvent |
R |
R1 |
Product |
Conversion (%) |
Yield (%) |
| Reaction conditions: compound 1 (0.1 mmol) was dissolved in the appropriate solvent (1.5 mL) in the presence of protected α-amino acids 19–26 (1.0 mmol) and treated with IBX (0.3 mmol) at 45 °C for 72 h. |
| 1 |
19 |
THF |
H |
H |
27(11) |
85 |
27(5) |
| 2 |
19 |
MeOH |
H |
H |
27(11) |
≥98 |
65(10) |
| 3 |
20 |
THF |
CH3 |
H |
28(11) |
≥98 |
48(10) |
| 4 |
20 |
MeOH |
CH3 |
H |
28(11) |
≥98 |
80(5) |
| 5 |
21 |
THF |
CH(CH3)2 |
H |
29(11) |
70 |
40(16) |
| 6 |
21 |
MeOH |
CH(CH3)2 |
H |
29(11) |
95 |
60(13) |
| 7 |
22 |
THF |
CH2CH(CH3)2 |
H |
30(11) |
87 |
43(21) |
| 8 |
22 |
MeOH |
CH2CH(CH3)2 |
H |
30(11) |
96 |
65(11) |
| 9 |
23 |
THF |
CH2C6H5 |
H |
31(11) |
≥98 |
90(8) |
| 10 |
23 |
MeOH |
CH2C6H5 |
H |
31(11) |
81 |
45(25) |
| 11 |
24 |
THF |
CH2CH2CH2 |
|
32(11) |
87 |
53(13) |
| 12 |
24 |
MeOH |
CH2CH2CH2 |
|
32(11) |
85 |
11(36) |
| 13 |
25 |
THF |
 |
H |
33(11) (35) |
≥98 |
80(4)(7) |
| 14 |
25 |
MeOH |
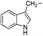 |
H |
33(11) |
≥98 |
32(20) |
| 15 |
26 |
THF |
CH2CH2SCH3 |
H |
34(11)(35) |
≥98 |
72(5)(9) |
| 16 |
26 |
MeOH |
CH2CH2SCH3 |
H |
34(11) |
96 |
40(20) |
In this latter case, we were not able to recognize any specific relationship between the steric hindrance of the side-chain and the yield of desired product. A different reaction pathway was observed with α-amino acids 24–26, in which case THF was the best reaction solvent (Table 2, entries 9–16). In some cases (e.g. Table 2, entries 3, 5, 14 and 16), the relatively low yield of desired product with respect to the high value of conversion of substrate can be due to formation of undesired side-products difficult to be detected and recovered from the reaction mixture. Note that in the case of the reactions performed with 25 and 26 in THF, a dimeric DOPA derivative (compound 35, Scheme 3) was detected in low amount as a side-product (7% and 9%, respectively). This compound showed all signals (and multiplicity) in 1H-NMR analysis (e.g. 7.46 ppm and 5.70 ppm singlets for symmetric couples of aromatic protons) expected for a dimeric structure, and was probably obtained by oxidative coupling of radical intermediates. This hypothesis is in accordance with previously reported results during IBX oxidation of 2-methoxy- and 2-methyl-substituted phenols in THF.40 The C(6)–C(6) regiochemistry in 35 was assigned by comparison of the 1H NMR multiplicity of aromatic protons with similar dimer species characterized in the polymerization of 3-(3,4-dihydroxyphenyl) propionic acid (DHPA) with Fe3+ ions.41
 |
| | Scheme 3 IBX mediated oxidative functionalization of N-Boc-Tyr, N-Boc-DOPA-OMe (15), N-Boc-Pro-N-Boc-DOPA-OMe (16), N-Boc-Trp-N-Boc-DOPA-OMe (17), N-Boc-Met-N-Boc-DOPA-OMe (18), from acceptable to high yield (Scheme 2, Table 1, entries 3–9). | |
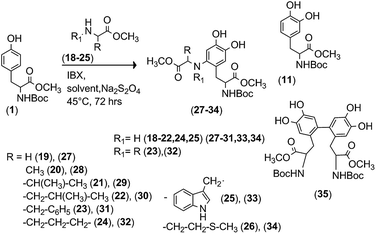 |
| | Scheme 4 IBX mediated oxidative functionalization of N-Boc-Tyr-OMe (1) with O-protected α-amino acids. | |
Finally, in view of possible large scale applications, we evaluated the use of heterogeneous conditions by applying polymer supported IBX-amide, that is an easily recoverable and reusable oxidant.42 The oxidation of Boc-Tyr-OMe (1) in the presence of Gly-OMe (19) in MeOH was performed under previously optimized experimental conditions. Comparable results in terms of conversion of substrate and yield of 27 were obtained with respect to IBX. Recycling experiments proceeded with success (Scheme 5). After the disappearance of compound (1), the IBX-amide was recovered by filtration, regenerated with Oxone® as reported43 and reused in further oxidations. As shown, IBX-amide was used for at least five cycles to give 27 without appreciable loss of efficiency (Table 3, runs 1–5).
 |
| | Scheme 5 Supported IBX-amide mediated oxidative functionalization of N-Boc-Tyr-OMe (1) with Gly-OMe (19). | |
Table 3 Reusability of heterogeneous IBX-amide in oxidative functionalization of N-Boc-Tyr-OMe (1) with Gly-OMea (19)
| Run |
Amino acid |
Product |
Conversion (%) |
Yield (%) |
| Reaction conditions: compound 1 (0.1 mmol) was dissolved in MeOH (1.5 mL) in the presence of protected α-amino acids 19 (1.0 mmol) and treated with IBX-amide at 45 °C for 72 h. |
| 1 |
19 |
27(11) |
≥98 |
65(8) |
| 2 |
19 |
27(11) |
≥98 |
67(9) |
| 3 |
19 |
27(11) |
≥98 |
63(10) |
| 4 |
19 |
27(11) |
≥98 |
65(11) |
| 5 |
19 |
27(11) |
≥98 |
66(7) |
3. Antioxidant activity
Radical oxygen centered species (ROS) are formed in the cell under normal metabolic and physiologic processes.44 In PD, hydroxyl and superoxide radicals, which are produced by oxidative phosphorylation,45 can damage mitochondrial DNA (mtDNA), causing modulation in the electron transport chain (ETC).46 The catechol pharmacophore in DOPA plays a key role in scavenging ROS by formation of stable phenoxyl radical species,47 as evaluated by standard and modified comet assays in mammalian cells.48
Furthermore, the administration of L-DOPA reduces hypoxia conditions and induces the over-expression of ORP150 (oxygen regulated protein 150 kDa) with concomitant cytoprotective effects, and possible activation of endogenous antioxidant mechanisms.49 On the basis of these data, we started to evaluate the antioxidant activity of novel synthesized DOPA peptidomimetic derivatives. The in vitro antioxidant activity of catecol compounds is usually determined by spectrophotometric analyses. We evaluated the 2,2-diphenyil picrylhydrazyl (DPPH) radical scavenging properties of compounds 10, 12–18 and 27–34, using 11 as reference. Briefly, the appropriate compound was added to freshly prepared DPPH solution (6 × 10−5 M in EtOH) and the decrease in absorbance (475 nm) was determined at different times until the reaction reached a plateau. The kinetic of the process was analyzed for each concentration tested, and the rate of DPPH remaining at the steady state was estimated. This value was used to calculate the IC50 (defined as the concentration of substrate that causes 50% loss of DPPH activity). Results for news O–C and N–C bonded L-DOPA peptidomimetics are reported in Fig. 1a and b respectively. All compounds showed appreciable antioxidant activity compared to DOPA. Note that glycine derivatives 10 and 27 showed the highest antioxidant activity. As a general trend, the IC50 decreased by increasing the steric hindrance of the side-chain substituent in the aliphatic amino acid family.
 |
| | Fig. 1 2,2-Diphenyil picrylhydrazyl (DPPH) radical scavenging properties of compounds 10, 12–18 and 27–34. (a) IC50 value of O–C bonded L-DOPA peptidomimetics 10, and 12–18 (b) IC50 value of N–C bonded L-DOPA peptidomimetics 27–34. IC50 is the drug concentration causing 50% inhibition of the desired activity. Each experiment was conducted in triplicate. | |
3.1. Evaluation of the genotoxic potential
The genotoxic potential of N-Boc-Gly-N-Boc-DOPA-OMe (10), N-Boc-Val-N-Boc-DOPA-OMe (13), Gly-N-Boc-DOPA-OMe (27) and Val-N-Boc-DOPA-OMe (29), selected as representative examples of couples of O–C and N–C bonded L-DOPA peptidomimetics, was evaluated in Chinese Hamster Ovary (CHO) cells, by analyzing the induction of chromosomal aberrations, which are highly predictive of long term genetic effects and cancer risk.50 Compounds like N-Boc-DOPA-OMe (11), and the natural L-DOPA peptides Boc-Gly-DOPA (36) and Boc-Val-DOPA (37) (Fig. 2), were also evaluated as reference compounds. The peptides 36 and 37 have been synthesized by selective oxidation of corresponding tyrosine containing substrates with native tyrosinase, as previously reported.51 All samples were prepared immediately before the analysis by solubilization of the appropriate compound in a small aliquot of dimethylsulphoxide (DMSO) followed by addition to the culture medium so that the final DMSO concentration did not exceed 1%. No precipitation was observed in the treatment medium at any dose level with any test compound. Following dose-range finding experiments, toxic dose-levels causing a complete suppression of mitotic activity and/or cell lethality were identified.
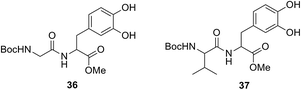 |
| | Fig. 2 Structure of L-DOPA peptides Boc-Gly-DOPA (36) and Boc-Val-DOPA (37). | |
The assay was then performed using a set of five dose-levels spaced by a factor of 1.8, starting from a maximum concentration expected to induce moderate toxicity, as evaluated by mitotic index (MI).52 All tested compounds, following a treatment of 24 hours, induced at the highest dose-levels selected, moderate reduction of mitotic indices up to 32–67% of the concurrent solvent controls (Tables 4 and 5). Notably, compound 13 proved to be the most active compound in terms of reduction of MI compared to references 11, 36 and 37, since active at lower concentrations (Table 5, entry 8 versus Table 4, entries 2, 8 and 14). Alternatively, compounds 10, 27 and 29, although less active than reference compound 11 (Table 5, entries 2, 14 and 20 versus Table 4, entry 2), showed a greater capability to reduce MI as compared with ref. 36 and 37 (Table 5, entries 2, 14 and 20 versus Table 4, entries 8 and 14). No statistically significant increases in the incidence of chromosomal aberrations were observed at any dose-level employed with any compound (Tables 4 and 5), indicating the absence of genotoxic potential.
Table 4 Analysis of mitotic index and chromosomal aberrations of reference compounds 11, 36 and 37, in CHO cells
| Entry |
Compound |
Dose-levels (μg mL−1) |
MIa (%) |
Relative MIb |
Aberrant cellsc (%) |
Stat. sig. |
| Mitotic indices (MI) corresponding to the ratio between the number of cells in a population undergoing mitosis to the number of cells not undergoing mitosis (interphase cells) out of a total of 1000 cells scored and expressed as percentage. Value of MI relative to solvent control. The solvent control value is considered to be equal to 100. Percentage of cells bearing aberrations (excluding gaps). |
| 1 |
Control 11 |
— |
6.9 |
100 |
4 |
— |
| 2 |
1.76 |
7.9 |
114 |
3 |
NS |
| 3 |
3.17 |
6.9 |
100 |
2 |
NS |
| 4 |
5.72 |
7.9 |
114 |
3 |
NS |
| 5 |
10.3 |
6.9 |
100 |
2 |
NS |
| 6 |
18.5 |
4.6 |
67 |
7 |
NS |
| 7 |
Control 36 |
— |
9.3 |
100 |
3 |
— |
| 8 |
9.91 |
8.8 |
95 |
2 |
NS |
| 9 |
17.8 |
7.7 |
83 |
3 |
NS |
| 10 |
32.1 |
4.5 |
48 |
2 |
NS |
| 11 |
57.8 |
3.7 |
40 |
2 |
NS |
| 12 |
104 |
3.2 |
34 |
3 |
NS |
| 13 |
Control 37 |
— |
9.3 |
100 |
3 |
— |
| 14 |
22.9 |
8.8 |
95 |
2 |
NS |
| 15 |
41.2 |
7.1 |
76 |
5 |
NS |
| 16 |
74.1 |
5.6 |
60 |
2 |
NS |
| 17 |
133 |
3.8 |
41 |
1 |
NS |
| 18 |
240 |
3.2 |
34 |
3 |
NS |
Table 5 Analysis of mitotic index and chromosomal aberrations of peptidomimetics 10, 13, 27 and 29, in CHO cells
| Entry |
Compound |
Dose-levels (μg mL−1) |
MIa (%) |
Relative MIb |
Aberrant cellsc (%) |
Stat. sig. |
| Mitotic indices corresponding to the ratio between the number of cells in a population undergoing mitosis to the number of cells not undergoing mitosis (interphase cells) out of a total of 1000 cells scored and expressed as percentage. Value of MI relative to solvent control considered equal to 100. Percentage of cells bearing aberrations (excluding gaps). |
| 1 |
Control 10 |
— |
9.2 |
100 |
4 |
— |
| 2 |
6.10 |
68.2 |
89 |
3 |
NS |
| 3 |
11.0 |
7.4 |
80 |
5 |
NS |
| 4 |
19.8 |
6.4 |
70 |
5 |
NS |
| 5 |
35.6 |
5.8 |
63 |
3 |
NS |
| 6 |
64.0 |
3.9 |
42 |
2 |
NS |
| 7 |
Control 13 |
— |
9.2 |
100 |
4 |
— |
| 8 |
1.33 |
8.4 |
91 |
2 |
NS |
| 9 |
2.40 |
7.7 |
84 |
2 |
NS |
| 10 |
4.32 |
6.2 |
67 |
3 |
NS |
| 11 |
7.78 |
4.2 |
46 |
4 |
NS |
| 12 |
14.0 |
3.7 |
40 |
4 |
NS |
| 13 |
Control 27 |
— |
9.2 |
100 |
4 |
— |
| 14 |
4.57 |
9.7 |
105 |
2 |
NS |
| 15 |
8.23 |
8.0 |
87 |
2 |
NS |
| 16 |
14.8 |
7.3 |
79 |
5 |
NS |
| 17 |
26.7 |
4.5 |
49 |
4 |
NS |
| 18 |
48.0 |
4.1 |
45 |
3 |
NS |
| 19 |
Control 29 |
— |
6.9 |
100 |
4 |
— |
| 20 |
6.29 |
6.9 |
100 |
5 |
NS |
| 21 |
11.3 |
6.1 |
88 |
3 |
NS |
| 22 |
20.4 |
5.7 |
83 |
2 |
NS |
| 23 |
36.7 |
4.8 |
70 |
1 |
NS |
| 24 |
66.0 |
3.1 |
45 |
3 |
NS |
3.2. Antioxidant activity in cultured mammalian cells
The antioxidant activity of peptidomimetics 10, 13, 27 and 29, and reference compounds 11, 36 and 37, was further evaluated in mouse lymphoma L5178Y (TK+/−) cells, in order to have a more realistic scenario of the complex interaction within the cell in biological systems. To this aim, the potential antioxidant activity in vivo of these compounds was assessed by their ability to reduce the extent of DNA breakage induced by hydrogen peroxide (H2O2) at 0.25 μM for 5 min, using a slightly modified version of the alkaline comet assay as previously proposed.53 The L5178Y mouse lymphoma cells were treated with the appropriate compound at the highest non cytotoxic concentration. For the reference compounds 11, 36 and 37 the selected dose-levels were 18.5, 104 and 240 μg mL−1, respectively, as shown in Table 4 (entries 6, 12 and 18), while for peptidomimetics (10, 13, 27 and 29) the selected dose-levels were 64, 14, 48 and 66 μg mL−1, respectively, as shown in Table 5 (entries 6, 12, 18, 24 respectively). No increase in DNA migration was noted after treatment with any of tested compounds when present alone, while marked and statistically significant increases in tail moment values, reflecting increased DNA breakage, were observed in the H2O2 treated cells.
The highest protection against DNA breakage was observed for N-Boc-DOPA-OMe (11) and L-DOPA peptides Boc-Gly-DOPA (36) and Boc-Val-DOPA (37), which were able to reduce the tail moment values (TM) in the range of 94–96% (Table 6, entries 2–4). The TM is defined as the product of the tail length and the fraction of total DNA in the tail. This data incorporates a measure of both the smallest detectable size of migrating DNA (reflected in the comet tail length) and the number of relaxed/broken DNA fragments (represented by the intensity of DNA in the tail). A lower but still significant protection was also observed for O–C and N–C bonded L-DOPA peptidomimetics, N-Boc-Val-N-Boc-DOPA-OMe (13), Val-N-Boc-DOPA-OMe (29) and N-Boc-Gly-N-Boc-DOPA-OMe (10) (Table 6, entries 5–7). The Gly-N-Boc-DOPA-OMe (27) proved to be the less active compound (Table 6, entry 8). The results presented refer to a third trial performed which strictly confirmed previous finding for which results have not been reported. On the basis of these data, the modification of the nature of the bond between amino acid residues (that is, natural peptide bond linkage versus modified O–C and N–C bonds) does not alter in a significant way the antioxidant activity of the compounds in L5178Y (TK+/−) mouse lymphoma cells. However, L-DOPA peptides are still the most active. This trend could reasonably be due to modification of stereo-electronic properties of the catecholic moiety in peptidomimetic derivatives, following the functionalization of the aromatic ring. For example, it is well known that the addition of oxygen atom (as OH group) on the catechol moiety significantly tune the value of the redox potential of the molecule.54 Moreover, in the family of peptidomimetic derivatives, the valine derivatives (compounds 13 and 29) showed the highest antioxidant activity. Representative image of TM reduction are shown in S2#.†
Table 6 Mean tail moment values (TM) + standard deviations (SD) of compounds 10, 11, 13, 27, 36 and 37 by the comet assay
| Entry |
Compound |
TMb |
TMc |
Reduction of TMa (%) |
| Reduction of the extent of DNA breakage induced by hydrogen peroxide (H2O2) at 0.25 μM for 5 min in L5178Y (TK+/−) mouse lymphoma cells. Experiment performed without H2O2. Experiment performed with H2O2. |
| 1 |
DMSO |
0.10 ± (0.43) |
17.14 ± (18.27) |
— |
| 2 |
36 |
0.25 ± (0.65) |
0.76 ± (1.37) |
96 |
| 3 |
37 |
0.22 ± (0.58) |
0.84 ± (1.15) |
95 |
| 4 |
11 |
0.80 ± (1.05) |
1.02 ± (2.08) |
94 |
| 5 |
13 |
0.62 ± (1.46) |
4.20 ± (5.03) |
75 |
| 6 |
29 |
0.22 ± (0.44) |
5.57 ± (4.75) |
68 |
| 7 |
10 |
0.38 ± (1.50) |
9.30 ± (7.81) |
46 |
| 8 |
27 |
0.42 ± (1.13) |
10.85 ± (11.09) |
36 |
4. Conclusions
Inspired to natural oxidative polymerization of tyrosine, a large panel of peptidase resistant L-DOPA peptidomimetics have been prepared in a selective way, using IBX as primary oxidant. The reaction was extended to heterogeneous conditions by applying supported IBX-amide reagent. Under these experimental conditions, two families of L-DOPA peptidomimetics were obtained, differing in the nature of the connection between the L-DOPA moiety and the appropriate α-amino acid residue, that is O–C versus N–C bonds. The regiochemistry of the addition between nucleophilic α-amino acid and electrophilic DOPA-quinone intermediate followed a Michael-like 1-4 selectivity. The novel L-DOPA peptidomimetics showed a significant antioxidant activity in 2,2-diphenyl picrylhydrazyl (DPPH) assay, confirming the “in vitro” radical scavenging capacity. In this latter case, compounds containing residues of glycine showed an antioxidant activity higher than L-DOPA, as a reference. It is interesting to note, that a different behavior was observed during the analysis of the antioxidant activity by the comet assay in L5178Y (TK+/−) mouse lymphoma cells, in which case valine derivatives were the most active compounds. These data further confirm the possible difference in the evaluation of the antioxidant activity between spectroscopic procedures and cellular models. In fact, cellular models better account of different aspects of cell uptake, distribution and toxicity.54 The antioxidant properties of novel L-DOPA peptidomimetics in L5178Y (TK+/−) mouse lymphoma model, were probably related to ROS scavenging mechanism rather than possible modulatory effect on the induced cellular DNA repair, since the time lapse between treatment with H2O2 and processing of cells for comet assay was approximately 10 min, a time clearly insufficient for DNA repair events to take place. Finally, the comparison of comet assay data between valine and glycine derivatives suggests a benign role of longer alkyl side chain substituent in the antioxidant activity.
5. Experimental
5.1. Materials
All solvents and reagents used were of analytical grade and were purchased from Aldrich Chemical Co. Silica gel 60 F254 plates and silica gel 60 were furnished from Merck. IBX was prepared in laboratory as described in the literature. Supported IBX-amide, 2,2-diphenyil-picrylhydrazyl (DPPH), sodium sulfate anhydrous (Na2SO4), Boc-Tyrosine-OMe (BTO; 1), and protected amino acids were purchased from Sigma-Aldrich.
5.2. General procedure for preparation of L-DOPA peptidomimetics
BTO 1 0.1 mmol was dissolved in 1.5 mL of the appropriate solvent, then 1.0 mmol of amino acid (2–9, 19–26) and 0.3 mmol of IBX were added. The reaction mixture was stirred at 45 °C for 72 h. The reaction was monitored by thin layer chromatography (TLC, n-hexane/EtOAc = 2.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.0). After the disappearance of substrate, the reaction mixture was treated with 2 mL of H2O and 2.0 eq. of Na2S2O4 stirring for 15 min. Then was added 2 mL of saturated solution of NaHCO3 and stirring for 30 min. The mixture was extracted several time with AcOEt and separated from H2O. The organic layer were collected dried with Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash-chromatography. Elemental analyses were performed with Perkin Elmer 2400 Series 2 CHNO/S apparatus. 1H and 13C NMR spectra were recorded on a Bruker (400 MHz) spectrometer. Mass spectra were recorded on a VG 70/250S spectrometer with an electron beam of 70 eV. Spectroscopic data are reported below. 1H and 13C NMR spectra are shown in S1#.† Rotatory values are calculated using a Perkin Elmer 343 Polarimeter. Analyses were performed in CHCl3.
:1.0). After the disappearance of substrate, the reaction mixture was treated with 2 mL of H2O and 2.0 eq. of Na2S2O4 stirring for 15 min. Then was added 2 mL of saturated solution of NaHCO3 and stirring for 30 min. The mixture was extracted several time with AcOEt and separated from H2O. The organic layer were collected dried with Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash-chromatography. Elemental analyses were performed with Perkin Elmer 2400 Series 2 CHNO/S apparatus. 1H and 13C NMR spectra were recorded on a Bruker (400 MHz) spectrometer. Mass spectra were recorded on a VG 70/250S spectrometer with an electron beam of 70 eV. Spectroscopic data are reported below. 1H and 13C NMR spectra are shown in S1#.† Rotatory values are calculated using a Perkin Elmer 343 Polarimeter. Analyses were performed in CHCl3.
5.2.1 N-Boc-Gly-N-Boc-DOPA-OMe (10). (34 mg, 70%), [α]20D = +10.1, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28–1.44 (18H, d, 6 × CH3), 2.97–3.00 (2H, m, CH2), 3.74 (3H, s, OCH3), 3.94 (2H, s, CH2) 4.62–5.14 (1H, m, CH), 7.24 (1H, s, CH), 7.42 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.08 (3 × CH3), 28.70 (3 × CH3), 32.47 (CH2), 42.89 (CH2) 52.47 (OCH3), 55.79 (CH), 79.53 (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) ), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (C
), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 485 [M + H] (100), 429 [M + H − 56] (43), 385 [M + H − 100] (22); elemental analysis calcd: C, 54.54; H, 6.66; N, 5.78; O, 33.02. Elemental analysis found: C, 54.51; H, 6.62; N, 5.73; O, 33.01.
O), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 485 [M + H] (100), 429 [M + H − 56] (43), 385 [M + H − 100] (22); elemental analysis calcd: C, 54.54; H, 6.66; N, 5.78; O, 33.02. Elemental analysis found: C, 54.51; H, 6.62; N, 5.73; O, 33.01.
5.2.2 N-Boc-DOPA-OMe (11). [α]20D = +7.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28–1.44 (18H, d, 6 × CH3), 2.96–3.03 (2H, m, CH2), 3.74 (3H, s, OCH3), 4.62–5.14 (1H, m, CH), 6.55 (1H, d, CH) J = 4, 6.69 (1H, s, CH), 6.79 (1H, d, CH) J = 4. 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.08 (3 × CH3), 32.47 (CH2) 52.47 (OCH3), 55.79 (CH), 79.53 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 171.90 (CO), MS (EI): m/z 312 [M + H] (100), 256 [M + H − 56] (47), 212 [M + H − 100] (24); elemental analysis calcd: C, 57.87; H, 6.80; N, 4.50; O, 30.83. Elemental analysis found: C, 57.81; H, 6.80; N, 4.50; O, 30.81.
5.2.3 N-Boc-Ala-N-Boc-DOPA-OMe (12). (33 mg, 65%), [α]20D = +23.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28–1.44 (18H, d, 6 × CH3), 1.50 (3H, s, CH3), 2.96–3.03 (2H, m, CH2), 3.74 (3H, s, OCH3), 4.12–4.14 (1H, m, CH), 4.62–5.14 (1H, m, CH), 7.24 (1H, s, CH), 7.42 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 17.20 (CH3) 28.08 (3 × CH3), 28.70 (3 × CH3), 32.47 (CH2), 52.47 (OCH3), 53.21 (CH) 55.79 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 499 [M + H] (100), 443 [M + H − 56] (37), 399 [M + H − 100] (19); elemental analysis calcd: C, 55.41; H, 6.87; N, 5.62; O, 32.09. Elemental analysis found: C, C, 55.40; H, 6.85; N, 5.61; O, 32.07.
5.2.4 N-Boc-Val-N-Boc-DOPA-OMe (13). (30 mg, 58%), [α]20D = +80.5, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 0.89–0.98 (6H, d, 2 × CH3) J = 4, 1.07–1.30 (18H, d, 6 × CH3), 2.14–2.20 (1H, m, CH), 2.96–3.03 (2H, m, CH2), 3.74 (3H, s, OCH3), 4.52–5.14 (1H, m, CH) 7.49 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 18.90 (2 × CH3) 28.08 (3 × CH3), 28.70 (3 × CH3), 30.41 (CH), 32.47 (CH2), 52.47 (OCH3), 55.79 (CH), 62.71 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 527 [M + H] (100), 471 [M + H − 56] (42), 427 [M + H − 100] (26); elemental analysis calcd: C, 57.02; H, 7.27; N, 5.32; O, 30.38. Elemental analysis found: C, 57.00; H, 7.23; N, 5.29; O, 30.35.
5.2.5 N-Boc-Leu-N-Boc-DOPA-OMe (14). (30 mg, 56%), [α]20D = +53.2, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 0.88–0.96 (6H, d, 2 × CH3) J = 4, 1.28–1.44 (18H, d, 6 × CH3), J = 4, 1.60–1.71 (2H, m, CH2), 2.10–2.21 (2H, m, 2 × CH), 2.96–3.03 (2H, m, CH2), 3.74 (3H, s, OCH3), 4.52–5.14 (1H, m, CH), 7.49 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 22.92 (2 × CH3), 24.83 (CH), 28.08 (3 × CH3), 28.70 (3 × CH3), 30.41 (CH), 32.47 (CH2), 40.57 (CH2) 52.47 (OCH3), 55.79 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 541 [M + H] (100), 485 [M + H − 56] (41), 441 [M + H − 100] (28); elemental analysis calcd: C, 57.76; H, 7.46; N, 5.18; O, 29.60. Elemental analysis found: C, 57.80; H, 7.41; N, 5.18; O, 29.62.
5.2.6 N-Boc-Phe-N-Boc-DOPA-OMe (15). (29 mg, 50%), [α]20D = +21.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.42–1.50 (18H, d, 6 × CH3), 2.92–3.10 (2H, m, CH2), 3.74 (3H, s, OCH3), 4.53–5.14 (1H, m, CH), 6.51 (2H, d, CH2), 6.71 (1H, s, CH), 6.79 (2H, d, CH2), 7.20 (1H, s, CH), 7.28 (1H, t, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.08 (3 × CH3), 28.70 (3 × CH3), 32.47 (CH2), 36.77 (CH2), 52.47 (OCH3), 55.79 (CH), 55.82 (CH) 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 125.01 (Car), 127.21 (Car), 129.41 (Car) 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 575 [M + H] (100), 519 [M + H − 56] (38), 475 [M + H − 100] (24); elemental analysis calcd: C, 60.62; H, 6.67; N, 4.88; O, 27.84. Elemental analysis found: C, 60.59; H, 6.61; N, 4.89; O, 27.82.
5.2.7 N-Boc-Pro-N-Boc-DOPA-OMe (16). (27 mg, 52%), [α]20D = +33.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.38–1.40 (18H, d, 6 × CH3), 1.60 (2H, m, CH2), 1.85 (2H, m, CH2), 3.35 (2H, m, CH2), 3.60 (2H, m, CH2), 3.73 (3H, s, OCH3), 4.31–4.50 (1H, m, CH), 6.45 (1H, s, CH), 6.90 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 24.07 (CH2), 28.02 (CH2), 28.08 (3 × CH3), 28.70 (3 × CH3), 32.47 (CH2), 52.47 (OCH3), 50.41 (CH), 55.79 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 525 [M + H] (100), 469 [M + H − 56] (48), 425 [M + H − 100] (29); elemental analysis calcd: C, 57.24; H, 6.92; N, 5.34; O, 30.50. Elemental analysis found: C, 57.21; H, 6.94; N, 5.33; O, 30.51.
5.2.8 N-Boc-Trp-N-Boc-DOPA-OMe (17). (31 mg, 51%), [α]20D = +41.5, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.42–1.50 (18H, d, 6 × CH3) J = 4, 2.92–3.09 (2H, m, CH2), 3.67 (3H, s, OCH3), 4.58–5.20 (1H, m, CH), 6.71 (1H, s, CH), 7.18 (2H, d, 2 × CH) 7.46 (1H, s, CH) J = 4, 7.38 (2H, t, 2 × CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.08 (3 × CH3), 28.40 (CH2) 28.70 (3 × CH3), 32.47 (CH2), 52.47 (OCH3), 55.79 (CH), 59.41 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 109.74 (Car), 111.14 (Car), 118.80 (Car), 119.31 (Car), 119.84 (Car), 121.10 (Car), 123.00 (Car), 127.44 (Car), 136.59 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 614 [M + H] (100), 558 [M + H − 56] (40), 514 [M + H − 100] (23); elemental analysis calcd: C, 60.67; H, 6.41; N, 6.85; O, 26.07. Elemental analysis found: C, 60.68; H, 6.38; N, 6.81; O, 26.17.
5.2.9 N-Boc-Met-N-Boc-DOPA-OMe (18). (32 mg, 57%), [α]20D = +61.1, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28–1.46 (18H, d, 6 × CH3) J = 4, 2.00–2.14 (2H, m, CH2), 2.06 (3H, s, S–CH3), 2.12–2.16 (2H, m, CH2), 2.91–2.94 (2H, t, CH2), 3.77 (3H, s, OCH3), 4.54–5.12 (1H, m, CH), 7.04 (1H, s, CH), 7.42 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 15.42 (CH3) 28.08 (3 × CH3), 28.70 (3 × CH3), 29.72 (CH2) 30.91 (CH2), 42.89 (CH2) 52.47 (OCH3), 56.59 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 559 [M + H] (100), 503 [M + H − 56] (43), 459 [M + H − 100] (27); elemental analysis calcd: C, 53.75; H, 6.86; N, 5.01; O, 28.64; S, 5.74. Elemental analysis found: C, 53.71; H, 6.84; N, 5.12; O, 28.70; S, 5.72.
5.2.10 Gly-N-Boc-DOPA-OMe (27). (26 mg, 65%); [α]20D = −14.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.44 (9H, s, 3 × CH3), 2.90–3.10 (2H, m, CH2), 3.74 (3H, s, OCH3), 3.93 (2H, s, CH2), 4.11 (3H, s, OCH3), 4.62–5.14 (1H, m, CH) J = 4, 7.24 (1H, s, CH), 7.42 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.04 (3 × CH3), 32.21 (CH2), 42.89 (CH2), 52.47 (OCH3), 52.59 (OCH3), 56.59 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 399 [M + H] (100), 343 [M + H − 56] (48), 299 [M + H − 100] (33); elemental analysis calcd: C, 54.26; H, 6.58; N, 7.03; O, 32.13. Elemental analysis found: C, 54.25; H, 6.59; N, 7.08; O, 32.13.
5.2.11 Ala-N-Boc-DOPA-OMe (28). (33 mg, 80%), [α]20D = −18.6, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.44 (9H, s, 3 × CH3), 2.29 (3H, s, CH3), 3.65–3.69 (2H, m, CH2), 4.67 (3H, s, OCH3), 4.75 (3H, s, OCH3), 4.79–4.83 (2H, m, CH), 5.19–5.39 (1H, m, CH), 7.02 (1H, s, CH), 7.54 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 18.07 (CH3), 28.04 (3 × CH3), 32.21 (CH2), 52.47 (OCH3), 52.59 (OCH3), 55.81 (CH), 56.59 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 142.34 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 413 [M + H] (100), 357 [M + H − 56] (45), 313 [M + H − 100] (29); elemental analysis calcd: C, 55.33; H, 6.84; N, 6.79; O, 31.03. Elemental analysis found: C, 55.30; H, 6.81; N, 6.76; O, 31.10.
5.2.12 Val-N-Boc-DOPA-OMe (29). (26 mg, 60%), [α]20D = −40.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 0.89 (6H, d, 2 × CH3) J = 4, 1.32 (9H, s, 3 × CH3), 2.90–3.10 (1H, m, CH), 3.74 (3H, s, OCH3), 3.76 (3H, s, OCH3), 3.81–3.86 (1H, m, CH) 7.49 (2H, s, 2 × CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 19.25 (2 × CH3), 28.04 (3 × CH3), 30.63 (CH), 32.21 (CH2), 52.47 (OCH3), 52.59 (OCH3), 55.81 (CH), 74.30 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 142.34 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 441 [M + H] (100), 385 [M + H − 56] (40), 341 [M + H − 100] (24); elemental analysis calcd: C, 57.26; H, 7.32; N, 6.36; O, 29.06. Elemental analysis found: C, 57.23; H, 7.31; N, 6.36; O, 29.05.
5.2.13 Leu-N-Boc-DOPA-OMe (30). (30 mg, 65%), [α]20D = −43.7, oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28 (9H, s, 3 × CH3), 1.32 (6H, d, 2 × CH3) J = 4, 2.61–2.64 (1H, m, CH), 2.95–3.03 (2H, m, CH2), 3.77 (3H, s, OCH3), 3.79 (3H, s, OCH3), 4.10 (2H, m, CH2), 4.61–5.03 (1H, m, CH) J = 4, 6.49 (1H, s, CH), 6.92 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 22.95 (2 × CH3), 28.04 (3 × CH3), 30.63 (CH), 32.21 (CH2), 40.75 (CH2), 52.47 (OCH3), 52.59 (OCH3), 55.81 (CH), 64.70 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 142.34 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 455 [M + H] (100), 399 [M + H − 56] (42), 355 [M + H − 100] (26); elemental analysis calcd: C, 58.14; H, 7.54; N, 6.16; O, 28.16. Elemental analysis found: C, 58.11; H, 7.57; N, 6.10; O, 28.16.
5.2.14 Phe-N-Boc-DOPA-OMe (31). (44 mg, 90%), [α]20D = −53.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm): 1.44 (9H, s, 3 × CH3), 3.04–3.06 (2H, m, CH2), 3.09–3.13 (2H, m, CH2), 3.70 (3H, s, OCH3), 3.77 (3H, s, OCH3), 4.28 (1H, m, CH), 5.24 (1H, m, CH), 6.46 (1H, s, CH), 6.81 (2H, t, 2 × CH), 6.94 (2H, d, 2 × CH) J = 4, 7.24 (1H, s, CH), 7.38 (2H, t, 2 × CH) J = 8. 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.04 (3 × CH3), 32.21 (CH2), 36.95 (CH2), 42.89 (CH2) 52.47 (OCH3), 52.59 (OCH3), 56.59 (CH), 69.69 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 124.92 (Car), 127.62 (Car), 129.57 (Car), 144.76 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 489 [M + H] (100), 433 [M + H − 56] (38), 389 [M + H − 100] (19); elemental analysis calcd: C, 61.46; H, 6.60; N, 5.73; O, 26.20. Elemental analysis found: C, 61.46; H, 6.61; N, 5.73; O, 26.27.
5.2.15 Pro-N-Boc-DOPA-OMe (32). (23 mg, 53%), [α]20D = +43.1, oil, 1H NMR (400 MHz, CDCl3): δH (ppm): 1.44 (9H, 6, 3 × CH3), 1.60 (2H, m, CH2), 1.85 (2H, m, CH2), 3.35 (2H, m, CH2), 3.60 (2H, m, CH2), 3.70 (3H, s, OCH3), 3.77 (3H, s, OCH3), 4.31–5.10 (1H, m, CH), 6.45 (1H, s, CH), 6.90 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 27.56 (CH2), 28.04 (3 × CH3), 29.81 (CH2) 32.28 (CH2), 42.89 (CH2) 52.47 (OCH3), 52.59 (OCH3), 56.59 (CH), 74.64 (CH), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 439 [M + H] (100), 383 [M + H − 56] (45), 339 [M + H − 100] (30); elemental analysis calcd: C, 57.52; H, 6.90; N, 6.39; O, 29.19. Elemental analysis found: C, 57.50; H, 6.86; N, 6.36; O, 29.16.
5.2.16 Trp-N-Boc-DOPA-OMe (33). (42 mg, 80%), [α]20D = −65.2, oil, 1H NMR (400 MHz, CDCl3): δH (ppm): 1.44 (9H, s, 3 × CH3), 3.68–3.73 (2H, m, CH2), 3.88–4.00 (2H, m, CH2), 4.10 (3H, s, OCH3), 4.15 (3H, s, OCH3), 4.28–5.14 (1H, m, CH), 5.06 (1H, s, CH), 6.71 (1H, s, CH), 7.18 (2H, d, 2 × CH), 7.46 (1H, s, CH) J = 4, 7.38 (2H, t, 2 × CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.04 (3 × CH3), 30.67 (CH2), 52.47 (OCH3), 52.59 (OCH3), 56.59 (CH), 70.70 (CH), 79.53 (–C), 80.48 (–C), 109.39 (Car), 111.21 (Car), 113.58 (CH), 117.31 (CH), 118.48 (Car), 119.31 (Car), 123.55 (Car), 127.66 (Car), 136.39 (Car), 142.34 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 527 [M + H] (100), 471 [M + H − 56] (43), 327 [M + H − 100] (27); elemental analysis calcd: C, 61.47; H, 6.30; N, 7.96; O, 24.26. Elemental analysis found: C, 61.41; H, 6.33; N, 7.92; O, 24.29.
5.2.17 Met-N-Boc-DOPA-OMe (34). (34 mg, 72%), [α]20D = −43.3, oil, 1H NMR (400 MHz, CDCl3): δH (ppm): 1.44 (9H, s, 3 × CH3), 2.38–2.40 (2H, m, CH2) J = 8, 2.72 (3H, s, S–CH3), 2.73–2.77 (2H, m, CH2), 2.99–3.03 (2H, m, CH2), 3.74 (3H, s, OCH3), 3.78 (3H, s, OCH3), 4.53–5.05 (1H, m, CH), 6.49 (1H, s, CH), 6.72 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 15.43 (CH3), 28.04 (3 × CH3), 31.68 (CH2), 32.10 (CH2), 32.21 (CH2), 52.47 (OCH3), 52.59 (OCH3), 56.59 (CH), 66.46 (CH2), 79.53 (–C), 80.48 (–C), 113.58 (CH), 117.31 (CH), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 158.32 (CO), 171.90 (CO), 172.64 (CO). MS (EI): m/z 473 [M + H] (100), 417 [M + H − 56] (41), 373 [M + H − 100] (26); elemental analysis calcd: C, 53.37; H, 6.83; N, 5.93; O, 27.09; S, 6.79. Elemental analysis found: C, 53.32; H, 6.80; N, 5.98; O, 27.08; S, 6.77.
5.2.18 N-Boc-DOPA-OMe-Boc-DOPA-OMe (35). Oil, 1H NMR (400 MHz, CDCl3): δH (ppm) = 1.28–1.44 (18H, d, 6 × CH3) J = 4, 2.91–3.05 (2H, m, CH2), 3.74 0 (3H, s, OCH3), 4.52–5.04 (1H, m, CH), 5.75 (1H, s, CH), 7.46 (1H, s, CH). 13C NMR (100 MHz, CDCl3): δH (ppm) = 28.08 (3 × CH3), 32.47 (CH2) 52.47 (OCH3), 55.79 (CH), 79.53 (–C), 113.58 (CH), 119.11 (Car), 119.31 (Car), 141.44 (Car), 144.76 (Car), 146.56 (Car), 155.80 (CO), 171.90 (CO), MS (EI): m/z 621 [M + H] (100), 565 [M + H − 56] (44), 521 [M + H − 100] (25); elemental analysis calcd: C, 58.06; H, 6.50; N, 4.51; O, 30.93. Elemental analysis found: C, 58.10; H, 6.53; N, 4.50; O, 30.93.
5.3. Test systems and culture conditions
L5178Y TK+/− clone (3.7.2C) mouse lymphoma cells were obtained from ATCC (CRL-9518™). Generation time, plating efficiency and absence of mycoplasma were checked at regular intervals. Stocks of the L5178Y cells are stored in liquid nitrogen and subcultures prepared from the frozen stocks for experimental use. Cells were grown in RPMI 1640 supplemented with 10% heat-inactivated horse serum, 2 mM L-glutamine and antibiotics (100 IU per mL penicillin and 100 IU per mL streptomycin) and incubated at 37 °C in a 5% carbon dioxide atmosphere and 100% nominal humidity. Chinese hamster ovary (CHO) cells were obtained from Prof. A. T. Natarajan (State University of Leiden, The Netherland). This cell line derives from the CHO isolated from an explant of the ovary of the Chinese hamster (Cricetulus griseus, 2n = 22). The CHO cell line is particularly useful for this kind of studies because of its stable karyotype (modal number is 21 chromosomes), short cell cycle (12–14 h) and its high plating efficiency. Stocks of CHO cells are stored in liquid nitrogen and subcultures are prepared from these stocks for experimental use. Cultures were grown as monolayer cultures in Ham's F-10 medium (Gibco BRL) supplemented with 15% foetal bovine serum, 4 mM L-glutamine and antibiotics (50 IU per mL penicillin and 50 IU per mL streptomycin). All incubations were at 37 °C in a 5% carbon dioxide atmosphere and 100% nominal humidity.
5.4. Chromosomal aberration assays
Approximately 24 hours before treatment exponentially growing cells were detached by trypsin action and an appropriate number of 25 cm2 plastic cell culture flasks containing 5 mL complete culture medium was individually inoculated with 3.0 × 105 cells. All samples were prepared immediately before the analysis by solubilization of the appropriate amount of compound in a small aliquot (500 mL) of dimethylsulphoxide (DMSO) followed by addition to the culture medium and successive dilution to obtain the desired dose-levels as reported in Tables 4 and 5 The final DMSO concentration did not exceed 1% to avoid possible side effects. No precipitation was observed in the treatment medium at any dose level with any tested compound. Test compound treatments of CHO cells were performed in the absence of a metabolic activation system for 24 hours (approximately 1.5 cell cycle). Colcemid at 0.27 mM was added during the last 3 hours of culture to accumulate cells in metaphase. Hypotonic shock was induced by 1% trisodium citrate solution for 10 minutes. Cell suspension was fixed in a mixture of methanol and glacial acetic acid (v/v 3:1) followed by three washes. Cytogenetic preparations for analyses of chromosomal aberrations and mitotic indices were stained with an aqueous solution of Giemsa (3%). For each experimental point 100 metaphases were scored for chromosomal aberrations and were classified according to the description of Savage. The mitotic index was expressed in percentage based on the number of metaphases present after a total of 1000 cells scored (interphases and metaphases). Solvent-treated cells served as negative control.
5.5. Comet assay
Cultures of mouse lymphoma cells at a concentration of 1 × 106 cells per mL were treated for 30 minutes at 37 °C in 5% carbon dioxide atmosphere and 100% nominal humidity, with each synthesized compound at a single dose-level, which was the highest concentration analyzed for scoring of chromosomal aberrations. In additional culture without any treatment which served as control was also included. At the end of treatment 10 μL of each cell suspension was added to 65 μL of 0.7% (w/v) low melting point agarose (Bio-Rad Lab.) and sandwiched between a lower layer of 1% (w/v) normal-melting agarose (Bio-Rad Lab.). For untreated cultured and culture treated compound, two sets of three slides each, were prepared. An aliquot of 50 μL of H2O2 (0.25 μM) was added to one set, while PBS was added to the parallel set. The slides were kept at +4 °C for 5 minutes and then immersed in lysing solution (2.5 M NaCl, 100 mM Na2EDTA, 10 mM Tris, pH 10) containing 10% DMSO and 1% Triton × 100 (ICN Biomedicals Inc.) at 4 °C overnight. Slides were then randomly placed in a horizontal gel electrophoresis apparatus with fresh alkaline electrophoresis buffer (300 mM NaOH, 1 mM Na2EDTA, pH > 13) and incubated for 25 min at 4 °C to allow for DNA unwinding and expression of alkali-labile sites. Electrophoresis was at 4 °C for 15 minutes at 30 V (1 V cm−1) and 300 mA. After electrophoresis, slides were immersed in 0.3 M sodium acetate in ethanol for 30 min. Slides were then dehydrated in an alcohol series (2 min at 70, 85, and 100%) and air-dried. Slides stained with 20 μg mL−1 ethidium bromide in the presence of antifade immediately before analysis, were examined at 40× magnification using an automated image analysis system (Comet Assay III; Perceptive Instruments, UK) connected to a fluorescence microscope (Zeiss Axioskop 2). DNA damage was quantified from the tail moment values. A number of 50 cells from each slide (150 cells in total) were analyzed per experimental point.
References
- M. C. Recio, J. Andujar and J. L. Rios, Curr. Med. Chem., 2012, 19, 2088 CrossRef CAS.
- B. D. Welch, A. P. van Demark, A. Heroux, C. P. Hill and M. S. Kay, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16828–16833 CrossRef CAS PubMed.
-
(a) L. D. Walensky, A. L. Kung and I. Escher, Science, 2004, 305, 1466–1470 CrossRef CAS PubMed;
(b) I. Avan, C. Dennis Hall and A. R. Katritzky, Chem. Soc. Rev., 2014, 43(10), 3575–3594 RSC.
- P. M. Abou-Sleiman, M. M. Muqit and N. W. Wood, Nat. Rev. Neurosci., 2006, 7, 207–219 CrossRef CAS PubMed.
- J. P. Bai and G. L. Amidon, Pharm. Res., 1992, 9, 969–978 CrossRef CAS.
- Y.-J. Fei, Y. Kanai, S. Nussberger, V. Ganapathy, F. H. Leibach, M. F. Romero, S. K. Singh, W. F. Boron and M. A. Hediger, Nature, 1994, 368(6471), 563–566 CrossRef CAS PubMed.
- K. Miyamoto, T. Shiraga, K. Morita, H. Yamamoto, H. Haga, Y. Taketani, I. Tamai, Y. Sai, A. Tsuji and E. Takeda, Biochim. Biophys. Acta, 1996, 1305, 34–38 CrossRef.
- C.-L. Wang, Y.-B. Fan, H.-H. Lu, T.-H. Tsai, M.-C. Tsai and H.-P. Wang, J. Biomed. Sci., 2010, 17, 1–8 CrossRef PubMed.
- H.-P. Wang, J.-S. Lee, M.-C. Tsai, H.-H. Lu and W. Hsu, Bioorg. Med. Chem. Lett., 1995, 5, 2195–2198 CrossRef CAS.
- J. O'Neill, F. Veitch and T. Wagner-Jauregg, J. Org. Chem., 1956, 21, 363–364 CrossRef.
- J. Niu and D. S. Lawrence, J. Biol. Chem., 1997, 272, 1493–1499 CrossRef CAS PubMed.
- S. K. Chowdhury, J. Eshraghi, H. Wolfe, D. Forde, A. G. Hlavac and D. Johnston, Anal. Chem., 1995, 67, 390–398 CrossRef CAS.
- S. Ranganathan and N. Tamilarasu, Tetrahedron Lett., 1994, 35, 447–450 CrossRef CAS.
- J. Ueda, T. Ozawa, M. Miyazaki and Y. Fujiwara, J. Inorg. Biochem., 1994, 55, 123–130 CrossRef CAS.
- J. C. Grammer, J. A. Loo, C. G. Edmonds, C. R. Cremo and R. G. Yount, Biochemistry, 1996, 35, 15582–15592 CrossRef CAS PubMed.
- F. Lazzaro, M. Crucianelli, F. de Angelis, V. Neri and R. Saladino, Tetrahedron Lett., 2004, 45, 9237–9240 CrossRef CAS PubMed.
- R. Saladino, M. Mezzetti, E. Mincione, A. T. Palamara, P. Savini and S. Marini, Nucleosides Nucleotides, 1999, 18, 2499–2510 CAS.
- G. Botta, M. Delfino, M. Guazzaroni, C. Crestini, S. Onofri and R. Saladino, ChemPlusChem, 2013, 78, 325–330 CrossRef CAS PubMed.
- C. Fonseca, M. R. M. Domingues, C. Simões, F. Amado and P. Domingues, J. Mass Spectrom., 2009, 44, 681–693 CrossRef CAS PubMed.
- R. Bernini, M. Barontini, F. Crisante, M. C. Ginnasi and R. Saladino, Tetrahedron Lett., 2009, 50, 6519–6521 CrossRef CAS PubMed.
- D. Magdziak, A. A. Rodriguez, R. W. van de Water and T. R. Pettus, Org. Lett., 2002, 4, 285–288 CrossRef CAS PubMed.
- A. Ozanne, L. Pouységu, D. Depernet, B. Francois and S. Quideau, Org. Lett., 2003, 5, 2903–2906 CrossRef CAS PubMed.
- S. Quideau, L. Pouységu, D. Deffieux, A. Ozanne, J. Gagnepain, I. Fabre and M. Oxobya, ARKIVOC, 2003, 6, 106–119 CrossRef.
- R. Bernini, S. Cacchi, G. Fabrizi and E. Filisti, Org. Lett., 2008, 10, 3457–3460 CrossRef CAS PubMed.
- R. Bernini, E. Mincione, M. Barontini and F. Crisante, J. Agric. Food Chem., 2008, 56, 8897–8904 CrossRef CAS PubMed.
- K. Marumo and J. Waite, Biochim. Biophys. Acta. Protein. Struct. Mol. Enzymol., 1986, 872, 98–103 CrossRef CAS.
- R. Bernini, E. Mincione, F. Crisante, M. Barontini and G. Fabrizi, Tetrahedron Lett., 2009, 50, 1307–1310 CrossRef CAS PubMed.
- P. Vlieghe, V. Lisowki, J. Mcertines and M. Khrestchatinsky, Drug Discovery Today, 2010, 15, 40–56 CrossRef CAS PubMed.
- S. M. Miller, R. J. Simon, S. Ng, R. N. Zucherman, J. M. Kerr and W. H. Moos, Drug Dev. Res., 1995, 35, 20–32 CrossRef CAS PubMed.
- B. Liu, L. Burdine and J. Thomas Kodadek, J. Am. Chem. Soc., 2006, 128(47), 15228–15235 CrossRef CAS PubMed.
- L. Burdine, T. G. Gillette, H. J. Lin and T. J. Kodadek, J. Am. Chem. Soc., 2004, 126, 11442–11443 CrossRef CAS PubMed.
- L. A. Burzio and J. H. Waite, Biochemistry, 2000, 39, 11147–11153 CrossRef CAS PubMed.
- S. Jus, I. Stachel, W. Schloegl, M. Pretzler, W. Friess and M. Meyer, et al., Mater. Sci. Eng. C, 2011, 31, 1068–1077 CrossRef CAS PubMed.
- M. L. Mattinen, R. Lantto, E. Selinheimo, K. Kruus and J. Buchert, J. Biotechnol., 2008, 133, 395 CrossRef CAS PubMed.
- G. Matheis and T. R. Whitaker, J. Food Biochem., 1984, 8, 137 CrossRef CAS PubMed.
- E. Valero, R. Varon and F. Garcıa-Carmona, Arch. Biochem. Biophys., 2003, 416, 218–226 CrossRef CAS.
- M. Jimenez, F. Garcia-Carmona, F. Garcia Canovas, J. L. Iborra, J. A. Lozano and F. Martinez, Arch. Biochem. Biophys., 1984, 235, 438–448 CrossRef CAS.
- M. Uyanik, T. Mutsuga and K. Ishihara, Molecules, 2012, 17, 8604–8616 CrossRef CAS PubMed.
- V. Satam, A. Harad, R. Rajule and H. Pati, Tetrahedron, 2010, 66(39), 7659–7706 CrossRef CAS PubMed.
- S. Quideau, L. Pouységu, D. Deffieux, A. Ozanne, J. Gagnepain, I. Fabre and M. Oxobya, ARKIVOC, 2003, IV, 106–119 CrossRef.
- D. E. Fullenkamp, D. G. Barrett, D. R. Miller, J. W. Kurutz and P. B. Messersmith, RSC Adv., 2014, 4, 25127–25134 RSC.
- M. S. Yusubova and V. V. Zhdankin, Mendeleev Commun., 2010, 20, 185–191 CrossRef PubMed.
- T. K. Achar, S. Maitia and P. Mal, RSC Adv., 2014, 4, 12834–12839 RSC.
- R. A. Floyd, Exp. Biol. Med., 1999, 222, 236–245 CAS.
- P. A. Serra, G. Esposito, P. Enrico, M. A. Mura, R. Migheli, M. R. Delogu, M. Miele, M. S. Desole, G. Grella and E. Miele, Br. J. Pharmacol., 2000, 130, 937–945 CrossRef CAS PubMed.
- M. Reale, M. Pesce, M. Priyadarshini, M. A Kamal and A. Patruno, Curr. Drug Targets: CNS Neurol. Disord., 2012, 11, 430–438 CrossRef CAS.
- X. Liu, N. Yamada, W. Maruyama and T. Osawa, J. Biol. Chem., 2008, 283, 34887–34895 CrossRef CAS PubMed.
- C. Lipinski, F. Lombardo, B. Dominy, P. Feeney, Cross Ref, CAS, Web of Science® Times Cited, 1997, 3107.
- S. K. Han, C. Mytilineou and G. Cohen, J. Neurochem., 1996, 66, 501–510 CrossRef CAS.
- A. V. Carrano, J. Occup. Med., 1986, 28, 1112–1116 CrossRef CAS PubMed.
- S. Derochette, T. Franck, A. Mouithys-Mickalad, G. Deby-Dupont, P. Neven and D. Serteyn, Intra- and extracellular antioxidant capacities of the new water-soluble form of curcumin (NDS27) on stimulated neutrophils and HL-60 cells, Chem.-Biol. Interact., 2013, 201, 49–57 CrossRef CAS PubMed.
- N. I. Fedotcheva, R. E. Kazakov, M. N. Kondrashova and N. V. Beloborodova, Toxicol. Lett., 2008, 180, 182–188 CrossRef CAS PubMed.
- M. Dusinská, Z. Dzupinková, L. Wsólová, V. Harrington and A. R. Collins, Mutagenesis, 2006, 21, 205–211 CrossRef PubMed.
- T. Francka, A. Mouithys-Mickalada, T. Robertc, G. Ghittic, G. Deby-Duponta, P. Nevend and D. Serteyna, Chem.-Biol. Interact., 2013, 206(2), 194–203 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09464j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.