
α-Lipoic acid protects HAECs from high glucose-induced apoptosis via decreased oxidative stress, ER stress and mitochondrial injury
Wenshuang Li,
Changyuan Wang,
Jinyong Peng,
Jing Liang,
Yue Jin,
Qi Liu,
Qiang Meng,
Kexin Liu and
Huijun Sun*
Department of Clinical Pharmacology, College of Pharmacy, Dalian Medical University, 9 West Section, Lvshun South Road, Lvshunkou District, Dalian, China 116044. E-mail: sunhuijun@dlmedu.edu.cn; Fax: +86 411 86110413; Tel: +86 411 86110413
First published on 13th August 2015
Abstract
α-Lipoic acid (LA) has a wide range of benefits in treating diabetes mellitus (DM) and DM vascular diseases, however, the specific mechanisms are not clearly understood. The purpose of this study was to investigate whether LA confers a cytoprotective effect on human aortic endothelial cells (HAECs) in the presence of high glucose concentration and the possible mechanisms involved in this effect. The apoptotic cells were detected by Annexin-V/PI and DAPI staining. Reactive oxygen species (ROS) were measured by flow cytometry assay. The change of mitochondrial transmembrane potential (ΔΨm) was analyzed by confocal laser scanning microscope. The expressions of NADPH oxidase 4 (Nox4), p22phox and Bcl-2 were measured by qRT-PCR and western blot analyses. Cytochrome C release from mitochondria, caspase-3 expression, endoplasmic reticulum (ER) stress and activation of nuclear factor-κB (NF-κB) were evaluated by western blot analyses. The results showed that LA inhibited endothelial cell apoptosis and decreased apoptosis related proteins expression. LA reduced ROS generation and over-expression levels of Nox4, p22phox induced by high glucose. LA also suppressed ER stress and the decreasing of ΔΨm as well as the activation of NF-κB. These results indicated the preventive effects of LA on HAECs apoptosis caused by high glucose and the effects might be obtained via inhibition of Nox4. These findings provide a new interpretation on the role of LA in the treatment of diabetes.
1. Introduction
Diabetes mellitus (DM) as a common metabolic endocrine disease, has become one of the most challenging health issues of the 21st century.1 Various acute and chronic DM complications have become an enormous threat to human health.2,3 The injury and apoptosis of endothelial cell is the earliest important event of DM vascular disease, and it is also the initial factor in the formation of atherosclerosis.4,5 Endothelial cell damage and apoptosis may destroy the endothelial monolayer and thereby lead to vascular injury. Therefore, the prevention of high-glucose induced endothelial cell injury and apoptosis is an important approach to treating DM complications.High concentration of glucose induces the reactive oxygen species (ROS) production in vascular endothelial cells.6 Also basal levels of ROS support cellular functions and act in intracellular signaling as second messengers after moderate increase over basal levels.7 However, drastically increased ROS levels such as hydrogen peroxide (H2O2) and superoxide radical (O2˙−) destroy the pro-oxidant/antioxidant balance to induced oxidative stress.7 Oxidative stress as a critical factor for many undesirable biological reactions, plays a key role in functional cells and the development of many diseases.8 The main sources of ROS include NADPH oxidase, lipooxygenase, xanthine oxidase and cyclooxygenase. These sources are interacting with each other continuously. However, NADPH oxidase is located at the center of these events.9 The NADPH oxidase family are multi-component protein complexes including Nox1-5, Duox1 and Duox 2. And in endothelial cells, Nox4 is responsible for the basal ROS production.10
Furthermore, an increasing number of studies have indicated that oxidative stress can disturb endoplasmic reticulum (ER) homeostasis and result in the accumulation of the unfolded and misfolded protein and pathological consequence, namely ER stress.11 In response to ER stress, the expression of ER stress-related proteins such as protein kinase R-like ER kinase (PERK), activation transcription factor 6 (ATF6), glucose-regulated protein (GRP78), C/EBP homologous protein (CHOP) was altered and leads to apoptosis.12–15 Apoptosis mediated by ER stress has been confirmed to be involved in the development of various diseases, including diabetes, cardiovascular and renal diseases.16,17
NF-κB is an oxidative stress-responsive transcription factor18 and transcriptional activity of NF-κB plays a critical role in endothelial dysfunction due to its sensitivity to ROS.19 NF-κB, as an inactive form existing in the cytoplasm, is stabilized by its inhibitory subunit I-κB. Stimulating the cells with H2O2, tumor necrosis factor (TNF) α and β, interleukin (IL)-1 and -2, dissociates I-κB from NF-κB, allowing free NF-κB from cytoplasm migrating to the nucleus, thereby transmitting multifarious signals.18 Hyperglycemia can promote the activation of NF-κB, which accelerates endothelial cell apoptosis.20 The mitochondrial injury was also found in high glucose treated endothelial cells.21 Persistent mitochondrial oxidative stress caused by ROS is known to regulate cytochrome C release from mitochondria to the cytosol22 and activates caspase-3,23 thereby triggering apoptosis.
High glucose induced endothelial apoptosis is a significant cause of diabetic vasculopathy, so seeking for new drugs which prevent high glucose induced endothelial cell apoptosis is a crucial therapeutic approach for diabetic complications. LA as a disulfide compound, has a powerful anti-oxidative effect by scavenging ROS thus protecting against oxidative stress induced apoptosis in some cell types.24,25 However, there is no information available on its mechanism involved in endothelial cell apoptosis caused by high glucose induced oxidative stress. The aim of the present study was to explore the protective effects and the potential mechanisms of LA on high glucose induced apoptosis in HAECs.
2. Materials and methods
2.1 Cell culture
Human aortic endothelial cells (HAECs) were purchased from Shanghai Bioleaf Bioteach Co., Ltd (Shanghai, China). The cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS) in a humidified incubator with 5% CO2 at 37 °C. One day before treatment, the culture medium was changed to DMEM medium without FBS. The experiments were carried out with five groups: one group remained in DMEM medium with a normal glucose concentration (11.5 mmol l−1, the C group); one group was cultured in DMEM medium with high glucose concentration (30 mmol l−1, the HG group); and three groups were cultured in DMEM medium with high glucose concentration (30 mmol l−1) supplemented with 50, 100, 200 μmol l−1 lipoic acid (the LA-L, LA-M, LA-H groups).2.2 Reagents and antibodies
LA was purchased from Jiangsu chemdrug co., ltd. (Jiangsu, China). D-Glucose was purchased from Beijing Solarbio science & technology co., ltd. (Beijing, China). DMEM medium was purchased from Gibco-BRL Company (Gaithersburg, MD, USA). Nox4 polyclonal antibody (Catalog no.: 14347-1-Ap), p65 polyclonal antibody (Catalog no.: 10745-1-Ap), Bcl-2 polyclonal antibody (Catalog no.: 12789-1-Ap), cleaved caspase-3 antibody (Catalog no.: AC033), cytochrome C polyclonal antibody (Catalog no.: 10993-1-Ap), ATF6 polyclonal antibody (Catalog no.: 24169-1-Ap), CHOP polyclonal antibody (Catalog no.: 15204-1-Ap) were all purchased from Proteintech Group Inc. P22phox was purchased from Santa Cruz Biotechnology Inc. (CA, USA). Rabbit anti-HSPA5 polyclonal antibody (Catalog no.: BA4293) was purchased from Wuhan boster bio-engineering limited company, Rabbit anti-PERK antibody (Catalog no.: bs-2468R) and Rabbit anti-phospho-PERK (Catalog no.: bs-3330R) were purchased from Beijing Biosynthesis biotechnology co. LTD. I-κBα polyclonal antibody, mouse anti-beta actin monoclonal antibody, goat anti-mouse IgG-HRP and goat anti-rabbit IgG-HRP were purchased from Zhongshan Golden Bridge Biotechnology Co, Ltd (Beijing, China).2.3 Apoptosis detection
Cell apoptosis were detected by an Annexin V-FITC apoptosis detection kit according to manufacturer's instructions. After trypsinization, HAECs were washed with PBS for three times and resuspended in 500 μl Annexin V binding buffer (Biouniquer Technology CO, LTD). Then, the cells were incubated in the dark with 5 μl Annexin V-FITC and 5 μl propidium iodide for 10 minutes at room temperature. The samples were analyzed by flow cytometry.2.4 Analysis of ROS generation
HAECs were incubated with high glucose and 50, 100, 200 μmol l−1 LA for 72 hours. After incubation, the cells were incubated with H2DCFHDA for 30 min at 37 °C. Then washed with PBS for 2 times and resuspended in PBS. The level of intracellular was measured using BD FACS Calibur Flow Cytometer (Becton, Dickinson and Company, USA).2.5 Analysis of mitochondrial transmembrane potential (ΔΨm)
After treatment with different concentrations of LA (50, 100, 200 μmol l−1) for 72 h, the HAECs were incubated with Rh123 (1 mg ml−1 in dimethyl sulfoxide) at 37 °C for 30 min and washed three times with PBS. The cells were harvested and analyzed by confocal laser scanning microscope (Leica microsystems).2.6 GSH and LDH analyses
In order to investigate the level of oxidative damage, glutathione (GSH) and LDH were measured. Briefly, HAECs were cultured in 24-well plates at a density of 2 × 104 per well and allowed to grow to desired confluence. The cells were then treated with high glucose supplemented with various concentrations of lipoic acid. Following 72 h of incubation, the cells were collected and measured according to the manufacturer's instructions (Beyotime, Nanjing, China).2.7 Western blot analysis
Equal amounts of proteins from cell extracts were subjected to western blot analysis on 8–10% sodium dodecyl sulfate (SDS)-polyacrylamide gels as described previously. Polyvinyldifluoride (PVDF) membranes were probed with primary antibody and then incubated with HRP-conjugated secondary antibody. The immunoreactive proteins were detected by chemiluminescence (ECL Plus, Beyotime Institute of Biotechnology, Shanghai, China) as described previously.26 β-Actin was used as internal control parameter.2.8 Quantitative real-time reverse transcription-PCR (qRT-PCR)
Total RNA from cells was isolated using RNAiso Plus (TaKaRa, Dalian, China) according to the manufacturer's instructions. qRT-PCR amplification and detection were performed using the 7500 qPCR system (Applied Biosystems, USA) with SYBR green (TaKaRa, Dalian, China). β-Actin was used as internal control and the 2(−delta delta CT) method was used to analyze the relative expression levels. The primer sequences were specific to human Nox4 (F 5′-GCTGCATCAGTCTTAACCGAAC-3′, R 5′-GGCTCTTCCATACAAATCTTCACA-3′); p22phox (F 5′-CAGTGGTACTTTGGTGCCTACTCC-3′, R 5′-GGTGGAGCCCTTCTTCCTCT-3′); Bcl-2 (F 5′-AGGAAGTGAACATTTCGGTGAC-3′, R 5′-GCTCAGTTCCAGGACCAGGC-3′).2.9 Transfection of small interference RNA
HAECs were transfected with Nox4 small interfering RNA (siRNA). The sense and anti sense strands of the Nox4 siRNA were 5′-CCAUGUGCCGAACACUCUUTT-3′ and 5′-AAGAGUGUUCGGCACAUGGT-3′. Human Nox4-specific siRNA was composited by Life Technologies (CA, USA). Briefly, the cells were plated in 6-well plates overnight and transferred to 1.5 ml DMEM medium without fetal bovine serum. Then, Nox4 siRNA duplexes and 5 μl of Lipofectamine™ 2000 (Life Technologies CA, USA) were mixed in 500 μl of DMEM medium for 20 min. We added the mixture to each well and incubated in a humidified incubator with 5% CO2 at 37 °C. 5 hours later, 1 ml DMEM medium containing 10% fetal bovine serum was added to each well. For experiments, HAECs were transferred with Nox4 siRAN for 24 hours and then treated with or without high glucose and LA for 72 hours.2.10 Statistical analysis
The data were analyzed by one-way ANOVA with LSD (least significant difference) post hoc test. Data are expressed as means ± SEM. The results were carried out at least three independent experiments. All statistical analysis was performed using SPSS 19.0 software. Where P < 0.05 was considered to be statistically difference.3. Results
3.1 LA inhibited high glucose induced apoptosis of HAECs
The apoptosis of endothelial cells is thought to play an important role in pathogenesis of cardiovascular disease, which is the foremost complication of diabetes.27 To quantitatively determine the anti-apoptotic effects of LA, the apoptosis of HAECs were measured by Annexin-V/PI staining after treatment with high glucose for 72 h. Fig. 1A indicated that rates of cell apoptosis were markedly increased in the high glucose group compared with the control group (P < 0.01). In contrast, LA at 50, 100, 200 μM significantly attenuated the apoptotic rate of the HAECs exposed to high glucose. Consistent with the result tested by Annexin-V/PI staining, DAPI staining analysis also demonstrated that normal HAECs exhibited homogeneous fluorescence intensity of nuclei, while heterogeneous intensity and chromatin condensation of nuclei appeared in high-glucose induced cells. And this effect induced by high glucose was markedly inhibited by LA (Fig. 1B). | ||
| Fig. 1 LA inhibited high glucose induced apoptosis of HAECs. (A) The level of apoptosis was measured by flow cytometry analysis. (B) Fluorescence microscopy images of AV/PI-stained HAECs. Apoptotic nuclei are shown by arrows (×20). Results represent the mean ± SEM of three independent experiments. ##p < 0.01 vs. control, **p < 0.01 vs. HG. | ||
3.2 LA enhanced the level of GSH, the expression of Bcl-2 and reduced cytochrome C and LDH release as well as the expression of cleaved caspase-3
It is well known that the GSH redox cycle represents the most important antioxidant system in endothelial cells and the depletion of intracellular GSH results in oxidative stress which is a known inducer of the transcription of specific genes involved in cell death.28 The intracellular level of GSH was measured in our present study in order to evaluate whether LA protects HAECs from injury by high glucose. The result showed that high glucose decreased GSH level compared to control group (P < 0.01). On the contrary, LA significantly increased the level of GSH compared to HG group in a concentration dependent manner (Fig. 2A). LDH, leaked from cells after plasma membrane disruption, is an indicator of irreversible cell death.29 As shown in Fig. 2B, the increased level of LDH induced by HG was significantly decreased by LA. We also noted the expression changes of anti-apoptotic protein Bcl-2 and mitochondrial cytochrome C release as well as pro-apoptotic protein cleaved caspase-3 by western blot and qRT-PCR (Fig. 2C–F). As a gene located at chromosome and encoded the 26-kD protein, Bcl-2 blocks the programmed cell death without affecting cellular proliferation. The mitochondrial dependent mechanism of caspase activation depends on the Bcl-2 family of proteins, including anti-apoptotic Bcl-2 which regulates mitochondrial outer membrane permeabilization.30 The results showed that Bcl-2 mRNA and protein expression level were decreased in the HG group compared with control group (p < 0.05, p < 0.01). Compared to the high glucose treatment alone, the Bcl-2 expression was significantly increased in the LA low, middle and high concentration groups (p < 0.05, P < 0.01). Cytochrome C, as one of the main apoptotic protease activating factors secreted by mitochondria, plays a key role in the cell apoptosis process.31 Cytochrome C release was increased in HG group compared with control group (P < 0.01). But this effect was markedly reversed by treatment with LA. Rhodamine 123 staining was used to measure the change in the fluorescence intensity as a measure of the mitochondrial membrane potential (ΔΨm), which drives the uptake and accumulation of Rh123 in the mitochondria. The hypofluorescence peak observed was indicative of a collapse in the ΔΨm and depolarization of the mitochondrial membrane. As shown in Fig. 2G, a collapse of the ΔΨm was observed in HAECs 72 h after treatment with high glucose, however, LA increased ΔΨm in a concentration dependent manner. At the same time, the expression of caspase-3, a representative caspase involved in response to ER stress and activated in cells undergoing apoptosis,32 was also enhanced by high glucose culture. However, LA could partially reverse the protein expression levels of cleaved caspase-3. These results suggested that LA could enhance anti-apoptotic protein expression and reduce pro-apoptotic protein expression and then play an anti-apoptosis role in HAECs treated with high glucose. | ||
| Fig. 2 LA enhanced the level of GSH, the expression of Bcl-2 and reduced cytochrome C and LDH release as well as the expression of cleaved caspase-3. (A) Effect of LA on the level of GSH. (B) Effect of LA on LDH release. (C) Effect of LA on Bcl-2 mRNA expression level induced by HG. (D) Effect of LA on Bcl-2 protein expression level induced by HG. (E) Effect of LA on cleaved caspase-3 expression induced by HG. (F) Effect of LA on level of cytochrome C release induced by HG. (G) Effect of LA on mitochondrial transmembrane potential (ΔΨm). β-Actin was used as internal control parameter. Results represent the mean ± SEM of three independent experiments. #p < 0.05 vs. control, ##p < 0.01 vs. control, *p < 0.05 vs. HG, **p < 0.01 vs. HG. | ||
3.3 LA attenuated high glucose induced ROS generation and NADPH oxidase over-expression
Hyperglycemia has been reported to increase ROS generation, which in turn activates cellular apoptotic pathways.33 In order to evaluate whether LA affects ROS generation, the level of intracellular ROS was measured using H2DCFDA. As shown in Fig. 3A, high glucose treatment alone significantly increased the ROS generation compared to control group (p < 0.01). Compared to the high glucose treatment alone, the ROS generation was significantly decreased in the LA low, middle and high concentration groups (p < 0.01). There results demonstrated that high glucose induced intracellular ROS production is significantly attenuated by LA in a concentration dependent manner. NADPH oxidase, an enzyme complex localized on the plasma membrane, is the major source of ROS. Among NADPH oxidase family, Nox4 is the mainly expressed form in endothelial cells. P22phox, as a docking protein for the other subunits which stabilizes NADPH oxidase, is required for all NADPH oxidase isoforms.34 Therefore, the effects of LA on high glucose induced Nox4 and p22phox mRNA and protein expressions were detected. High glucose treatment alone significantly increased mRNA level of Nox4 and p22phox by a factor of 10 and 8 fold over the control (p < 0.01), respectively (Fig. 3B and D). However, LA clearly inhibited Nox4 and p22phox mRNA over-expressions activated by high glucose. In terms of protein expression, high glucose markedly increased Nox4 and p22ophox expression by approximately 2 and 3-fold over that observed in control group (p < 0.01). The over expressions of Nox4 and p22ophox proteins induced by high glucose were also attenuated by LA (Fig. 3C and E).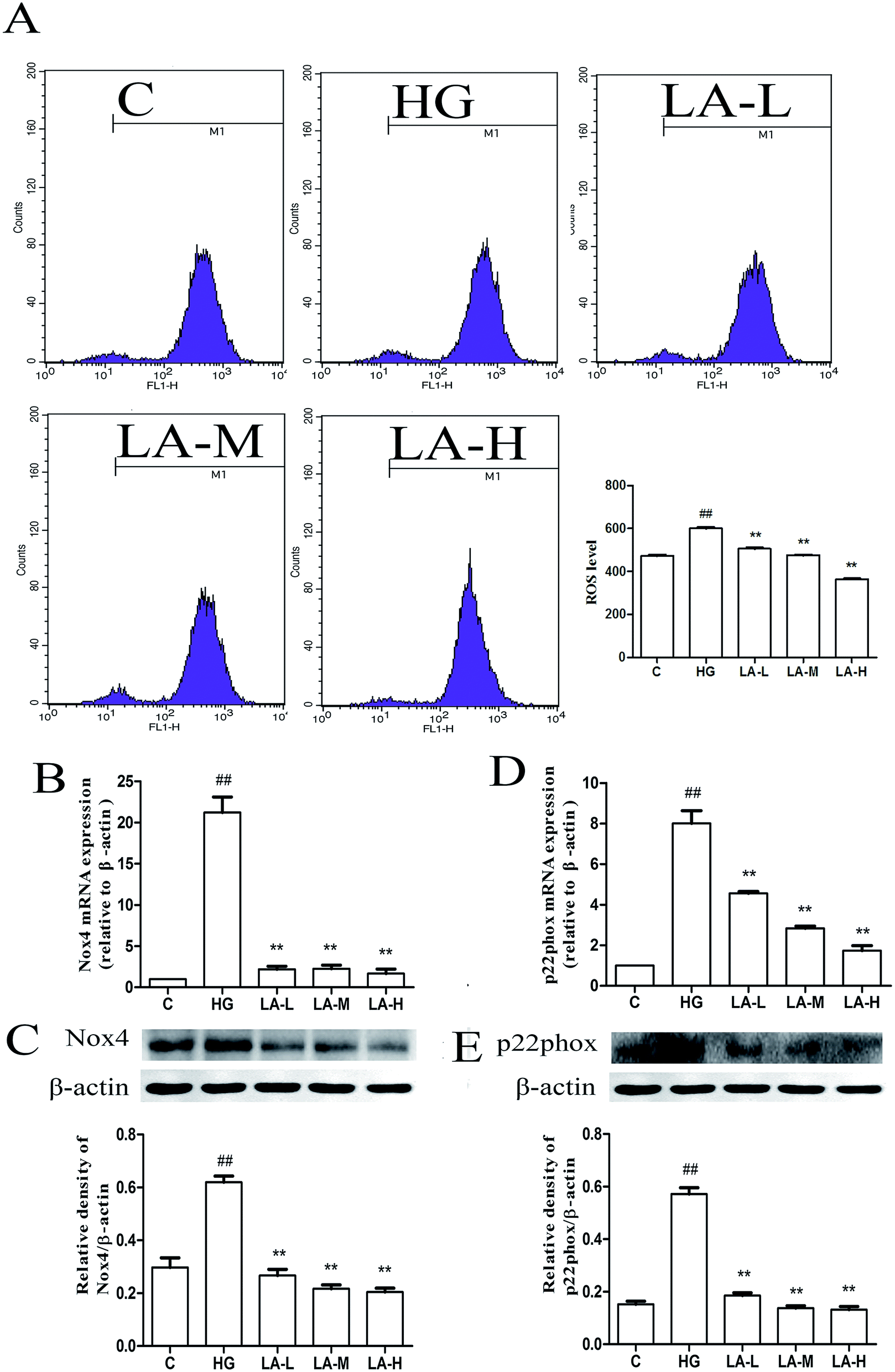 | ||
| Fig. 3 LA attenuated high glucose induced ROS generation and NADPH oxidase over-expression. (A) Effect of LA on ROS generation induced by HG. (B) Effect of LA on Nox4 mRNA expression level induced by HG. (C) Effect of LA on Nox4 protein expression level induced by HG. (D) Effect of LA on P22phox mRNA expression level induced by HG. (E) Effect of LA on P22phox protein expression level induced by HG. β-Actin was used as internal control parameter. Results represent the mean ± SEM of three independent experiments. ##p < 0.01 vs. control, **p < 0.01 vs. HG. | ||
3.4 LA attenuated high glucose induced ROS generation via inhibiting Nox4 in HAECs
In order to estimate whether the reduction of intracellular ROS production by LA was involved in the inhibition of Nox4, we employed siRNA to Nox4 and evaluated the high glucose induced ROS generation. In normal cells, Nox4 expression was significant. However, in Nox4 knock down cells, the expression of Nox4 was significantly inhibited in Fig. 4A and B. These results demonstrated that Nox4 was knocked down definitely. As shown in Fig. 4C, in normal cells, ROS production was increased in HG group compared to control group (p < 0.01). LA (100 μM) attenuated the high glucose induced ROS generation (p < 0.01). Moreover, in Nox4 knock down cells, the inhibition of LA on ROS generation was no longer observed. It is suggested that LA decreased ROS generation possibly via blocking Nox4. | ||
| Fig. 4 LA attenuated high glucose induced ROS generation and protected HAECs from apoptosis mainly by reducing Nox4 expression. (A) The Nox4 mRNA expression. (B) The Nox4 protein expression. (C) Effect of LA on ROS generation induced by HG in normal and Nox4 knock down cells. (D) The levels of apoptosis in normal and Nox4 knock down cells were measured by flow cytometry analysis. Results represent the mean ± SEM of three independent experiments. #p < 0.01 vs. control, ##p < 0.01 vs. control, **p < 0.01 vs. HG, &p < 0.05 vs. HG in Nox4 knock down cells. | ||
3.5 LA protected HAECs from apoptosis mainly by reducing Nox4 expression
In order to further identify whether LA protected HAECs from apoptosis induced by high glucose was involved in blocking Nox4, the total apoptotic rate was measured by Annexin-V/PI staining. As shown in Fig. 4D, in normal cells, the rate of apoptosis was increased in high glucose treated HAECs compared with control group (p < 0.01) and LA (100 μM) markedly decreased the apoptotic rate. In Nox4 knock down cells, the inhibition of LA on apoptotic rate was no longer observed. We also examined Bcl-2 and cleaved caspase-3 as well as mitochondrial cytochrome C release after transfection HAECs with Nox4 siRNA. As shown in Fig. 5A and B, in normal cells, Bcl-2 mRNA and protein expressions were decreased in high glucose treated group compared to control group (p < 0.01, p < 0.05). LA (100 μM) resulted in 3.3 and 5-fold rise compared to HG group (p < 0.01, p < 0.01). Moreover, in Nox4 knock down cells, LA increased Bcl-2 mRNA and protein expressions in 1.4 and 1.6-fold, respectively, compared to high glucose treatment alone (p < 0.01, p < 0.05). As shown in Fig. 5C, in normal cells, LA significantly decreased high glucose induced cleaved caspase-3 over-expressions in 1.7-fold (p < 0.01). Moreover, in Nox4 knock down cells, the significant inhibition of LA on cleaved caspase-3 expression induced by HG was no longer observed. As shown in Fig. 5D, in normal cells, LA decreased high glucose induced cytochrome C release in 1.6-fold decline (P < 0.01). In Nox4 knock down cells, LA resulted in 1.1-fold decline in mitochondrial cytochrome C release induced by high glucose. That is to say the extent of these decreases and increase in Nox4 knock down cells were lower than that in normal cells. It suggested that Nox4 mediated high glucose-induced apoptosis and LA increased Bcl-2 expression and decreased cytochrome C release as well as caspase-3 expression in high glucose treated HAECs partly through inhibiting Nox4 possibly.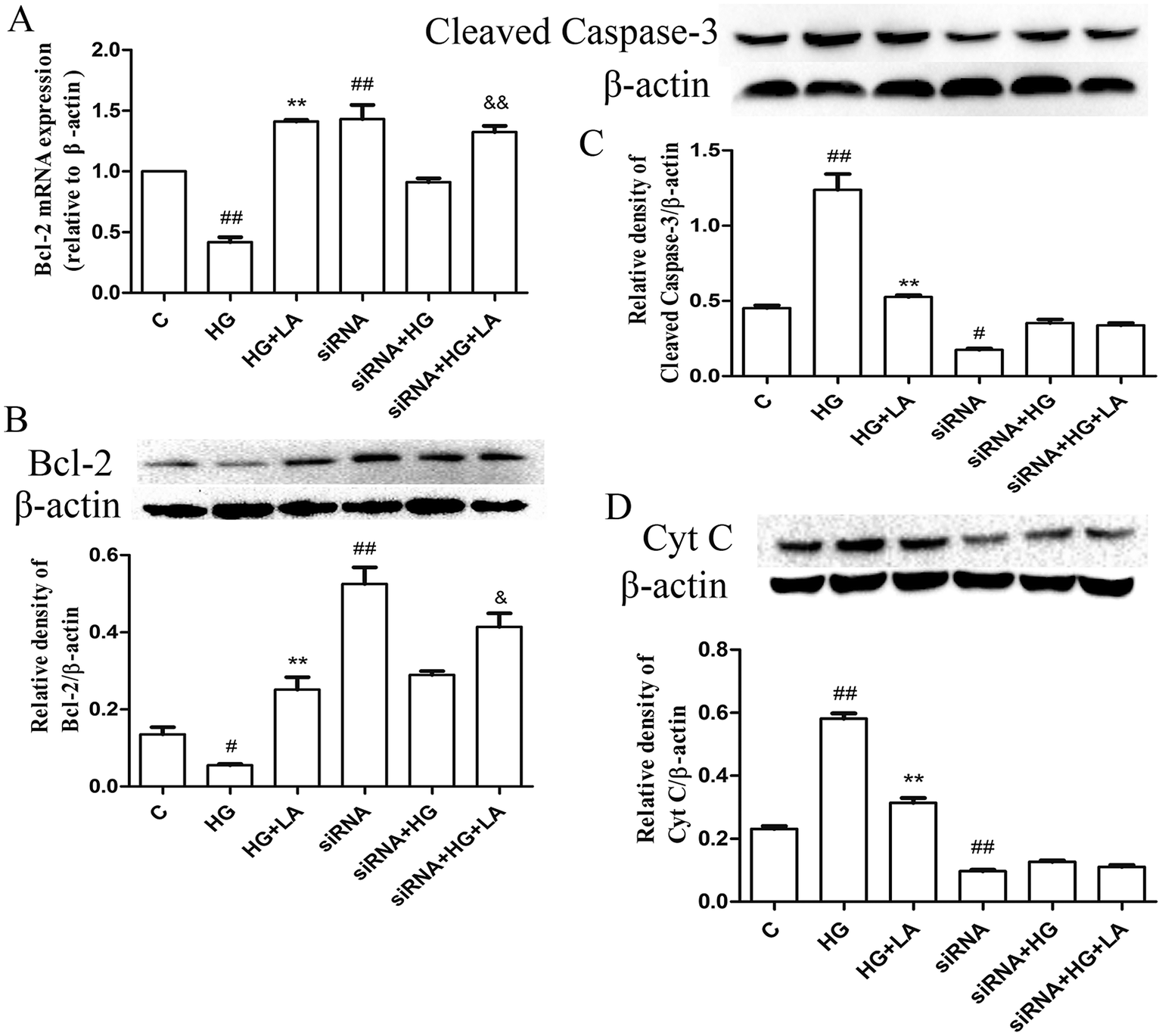 | ||
| Fig. 5 LA enhanced the expression of Bcl-2 and reduced cytochrome C release as well as the expression of cleaved caspase-3 by reducing Nox4 expression. (A) Effect of LA on Bcl-2 mRNA expression level induced by HG in normal and Nox4 knock down cells. (B) Effect of LA on Bcl-2 protein expression level induced by HG in normal and Nox4 knock down cells. (C) Effect of LA on caspase-3 expression level induced by HG in normal and Nox4 knock down cells. (D) Effect of LA on level of cytochrome C release induced by HG in normal and Nox4 knock down cells. β-Actin was used as internal control parameter. Data illustrated on the graph bar represent the mean ± SEM from three independent experiments. #p < 0.01 vs. control, ##p < 0.01 vs. control, **p < 0.01 vs. HG, &p < 0.05 vs. HG in Nox4 knock down cells. | ||
3.6 LA inhibited high glucose induced ER stress
Oxidative stress is known to stimulus ER stress.35 However, continuous ER stress stimulates apoptosis signal pathway and then results in cell apoptosis and tissue injury.36,37 To identify whether LA protected endothelial cells from high glucose induced apoptosis via ER stress, the expression levels of some proteins involved in ER stress pathway were analyzed by western blot. As depicted in Fig. 6A–D, high glucose caused an elevated expression level of ATF6, P-PERK, GRP78 and CHOP proteins after 72 hours treatment. However, treatment with LA could decrease over-expressions of GRP78, ATF6, P-PERK, and CHOP proteins in HAECs. Taken together, these data indicated that LA could relieve HAECs from high glucose induced ER stress. | ||
| Fig. 6 LA inhibited high glucose induced ER stress and NF-κB transcriptional activation in HAECs. (A) ATF6 protein level, (B) GRP78 protein level, (C) p-PERK protein level. (D) CHOP protein level. (E) I-κBα protein expression. (F) NF-κB/p65 protein expression. β-Actin or GAPDH was used as internal control parameter. Data illustrated on the graph bar represent the mean ± SEM from three independent experiments. ##p < 0.01 vs. control, *p < 0.05 vs. HG, **p < 0.01 vs. HG. | ||
3.7 LA inhibited high glucose induced I-κB degradation and NF-κB transcriptional activation in HAECs
NF-κB activation regulates cell death and cell viability and plays a key role in endothelial dysfunction resulting from its sensitivity to ROS.19 To examine the influence of LA on NF-κB activation, we examined I-κB degradation, p65 nuclear translocation by western blot (Fig. 6E and F). High glucose alone significantly induced I-κB in cytosol degradation. However, LA inhibited the high glucose reduced I-κB degradation in a concentration dependent manner. We then evaluated the translocation of p65 in the nucleus. High glucose significantly induced the translocation of p65, but treatment with LA could partly block high glucose induced p65 translocation. Accordingly, LA possibly suppressed NF-κB signaling pathway to produce anti-apoptosis effect.4. Discussion
ROS, as the most critical mediators of apoptotic signaling and subsequent cell death,6 have been well recognized to play important roles in the progression of pathologic lesions into cardiovascular disease (CVD).38,39In general, two ways of clearing free radicals induced damage are possible. The first way is by scavenging ROS through the induction of endogenous antioxidant systems. The second approach is to disturb the cause of oxidative stress via inhibiting enzymes that produce ROS. It is well known that the GSH as a free radical scavenger, has a vital role in endothelial cell defenses.40 In our present study, LA was shown to protect HAECs from oxidative stress induced injury via increasing the level of GSH. Thus, it may be assumed that the underlying mechanism behind the anti-apoptotic effects of LA may be due to its up-regulation of endogenous cellular anti-oxidant systems that are capable of scavenging ROS. Studies in the last decade have declared that NADPH oxidase family is the major source of ROS involved in redox signaling.41 In cardiovascular tissues, Nox4 is significantly more abundant than other Nox forms,9 while p22phox, a subunit functionally associated with Nox1-4, is a partner of ROS-generating systems.42 And p22phox is also known to stabilize and increase Nox4.34 In this study, we found that Nox4 and p22phox over-expressions induced by high glucose were both down-regulated by LA. In addition, using siRNA to Nox4, we demonstrated that LA decreased ROS generation possibly via blocking Nox4 as the second approach indicated.
The present study found that high glucose could markedly decrease Bcl-2 mRNA and protein expressions. In contrast, LA treatment significantly increased Bcl-2 mRNA and protein expressions. It is well reported that an anti-apoptotic protein Bcl-2 can suppress apoptosis through a mitochondria-dependent caspase pathway. Bcl-2 has been shown to prevent apoptosis stimulated by various stimuli, either by inhibiting the mitochondrial release of cytochrome C, which induce caspase activity, or by serving as an antioxidant.43 In resting state, as an electron shuttle in the mitochondrial respiratory chain, cytochrome C located in the mitochondrial intermembrane/intercristae spaces. In response to pro-apoptotic stimuli, mitochondrial outer membrane releases cytochrome C to the cytosol. In cytosol, cytochrome C mediates the activation of apoptotic protease activating factor 1, which is necessary for the proteolytic maturation of caspase-9 and caspase-3.31,44,45 Caspases family are synthesized as inactive pro-enzymes which are processed to active form in undergoing apoptotic cells.32 Therefore, these indicators were detected in our study to demonstrate that LA prevented HAECs from high glucose induced apoptosis through enhancing Bcl-2 expression and reducing cytochrome C release and caspase-3 expression. Furthermore, we knocked down Nox4 to investigate whether the effect of LA on mediating apoptosis and the expression of apoptosis-related proteins was related to its antioxidant effect. The results suggested that Nox4 mediated high glucose-induced apoptosis and the anti-apoptotic effect of LA in HAECs induced by high glucose possibly and partly via inhibiting Nox4.
Endoplasmic reticulum is sensitive to changes of intracellular homeostasis. Oxidative stress, the inhibition of protein glycosylation, a reduction in disulfide bond formation, calcium depletion and accumulation of unfolded proteins in the ER lumen may disrupt the ER function, and trigger the unfolded protein response (UPR), also known as “ER-stress”.46 Recently, ROS have emerged as important regulators of ER function and UPR activation in several diseased conditions.47 GRP78 protein was widely known as an ER stress marker. In this study, we found GRP78 protein was significantly increased in endothelial cells treated with high glucose. It is suggested that the ER stress signaling pathways were involved in high glucose-induced endothelial cell apoptosis. To protect against the negative influence of ER stress, ATF6 and PERK increase the capacity of ER on protein folding and degradation through attenuating protein translation, degenerating misfolded proteins and inducing molecular chaperons.48,49 CHOP is a downstream component of ER stress and it can be dramatically induced by ER stress, thereby, mediates apoptosis.50 The results of this study showed that high glucose induced activation of ER stress in HAECs could be effectively prevented by LA. Our result also indicated that LA decreased the activation of NF-κB in high glucose treated HAECs. ROS, along with other superoxide, is involved in the activation of NF-κB which is the redox-sensitive transcription factor.51 Researchers also demonstrated that the activation of NF-κB is dependent on NADPH oxidase-produced superoxide in endothelial cells.52 It is suggested that LA protected HAECs from oxidative stress induced apoptosis was partly via inhibiting NF-κB activation.
5. Conclusion
Our findings showed that LA prevented HAECs from high glucose induced apoptosis. And the protective effect of LA might be caused by its inhibitory effects on ROS generation, thereby, decreasing oxidative stress, ER stress and mitochondrial injury. Our findings will be helpful to find a new insight into the mechanism of LA in treating DM and the various acute and chronic DM complications.Conflict of interest
The authors report no declarations of interest.List of abbreviations
| ATF6 | Activation transcription factor 6 |
| CHOP C/EBP | Homologous protein |
| DM | Diabetes mellitus |
| ER | Stress endoplasmic reticulum stress |
| HAECs | Human aortic endothelial cells |
| GRP78 | Glucose-regulated protein 78 |
| LA | α-Lipoic acid |
| NF-κB | Nuclear factor-κB |
| Nox4 | NADPH oxidase 4 |
| PERK | Protein kinase R-like ER kinase |
| ROS | Reactive oxygen species |
| qRT-PCR | Quantitative real-time reverse transcription-PCR |
| UPR | Unfolded protein response |
Acknowledgements
This study was supported in part by Grants from the National Natural Science Foundation of China (No. 81273508) and Natural Science Fund of Science and Technology Bureau of Liaoning Province (No. 201102046).References
- T. Garabedian and S. Alam, Cardiovascular Diagnosis and Therapy, 2013, 3, 23–37 Search PubMed.
- M. M. Lui and M. Sau-Man, J. Thorac. Dis., 2012, 4, 164–172 Search PubMed.
- L. Salvado, L. Serrano-Marco, E. Barroso, X. Palomer and M. Vazquez-Carrera, Expert Opin. Ther. Targets, 2012, 16, 209–223 CrossRef CAS PubMed.
- R. M. Cubbon, N. Ali, A. Sengupta and M. T. Kearney, Curr. Vasc. Pharmacol., 2012, 10, 271–284 CrossRef CAS.
- Y. Yoshikawa and H. Yasui, Curr. Top. Med. Chem., 2012, 12, 210–218 CrossRef CAS.
- N. Toda and M. Nakanishi-Toda, Prog. Retinal Eye Res., 2007, 26, 205–238 CrossRef CAS PubMed.
- J. L. Martindale and N. J. Holbrook, J. Cell. Physiol., 2002, 192, 1–15 CrossRef CAS PubMed.
- M. Ott, V. Gogvadze, S. Orrenius and B. Zhivotovsky, Apoptosis, 2007, 12, 913–922 CrossRef CAS PubMed.
- A. Schramm, P. Matusik, G. Osmenda and T. J. Guzik, Vasc. Pharmacol., 2012, 56, 216–231 CrossRef CAS PubMed.
- Y. Zhou, H. Yan, M. Guo, J. Zhu, Q. Xiao and L. Zhang, Oxid. Med. Cell. Longevity, 2013, 2013, 374963 Search PubMed.
- M. Zettl, C. Adrain, K. Strisovsky, V. Lastun and M. Freeman, Cell, 2011, 145, 79–91 CrossRef CAS PubMed.
- Y. P. Yen, K. S. Tsai, Y. W. Chen, C. F. Huang, R. S. Yang and S. H. Liu, Arch. Toxicol., 2012, 86, 923–933 CrossRef CAS PubMed.
- T. H. Lu, C. C. Su, Y. W. Chen, C. Y. Yang, C. C. Wu, D. Z. Hung, C. H. Chen, P. W. Cheng, S. H. Liu and C. F. Huang, Toxicol. Lett., 2011, 201, 15–26 CrossRef CAS PubMed.
- F. Liu, K. Inageda, G. Nishitai and M. Matsuoka, Environ. Health Perspect., 2006, 114, 859–864 CrossRef CAS.
- S. Oyadomari, E. Araki and M. Mori, Apoptosis, 2002, 7, 335–345 CrossRef CAS.
- K. Srinivasan and S. S. Sharma, Life Sci., 2012, 90, 154–160 CrossRef CAS PubMed.
- B. Song, D. Scheuner, D. Ron, S. Pennathur and R. J. Kaufman, J. Clin. Invest., 2008, 118, 3378–3389 CAS.
- Z. Wei, Q. Peng, B. H. Lau and V. Shah, Gen. Pharmacol., 1999, 33, 369–375 CrossRef CAS.
- S. R. Kim, Y. H. Bae, S. K. Bae, K. S. Choi, K. H. Yoon, T. H. Koo, H. O. Jang, I. Yun, K. W. Kim, Y. G. Kwon, M. A. Yoo and M. K. Bae, Biochim. Biophys. Acta, Bioenerg., 2008, 1783, 886–895 CrossRef CAS PubMed.
- M. L. Sheu, F. M. Ho, R. S. Yang, K. F. Chao, W. W. Lin, S. Y. Lin-Shiau and S. H. Liu, Arterioscler., Thromb., Vasc. Biol., 2005, 25, 539–545 CrossRef CAS PubMed.
- D. Detaille, B. Guigas, C. Chauvin, C. Batandier, E. Fontaine, N. Wiernsperger and X. Leverve, Diabetes, 2005, 54, 2179–2187 CrossRef CAS.
- K. Sinha, J. Das, P. B. Pal and P. C. Sil, Arch. Toxicol., 2013, 87, 1157–1180 CrossRef CAS PubMed.
- B. Tharakan, F. A. Hunter, W. R. Smythe and E. W. Childs, Shock, 2008, 30, 571–577 CrossRef CAS PubMed.
- S. Balkis Budin, F. Othman, S. R. Louis, M. Abu Bakar, M. Radzi, K. Osman, S. Das and J. Mohamed, Rom. J. Morphol. Embryol., 2009, 50, 23–30 Search PubMed.
- X. Meng, Z. M. Li, Y. J. Zhou, Y. L. Cao and J. Zhang, Clin. Exp. Med., 2008, 8, 43–49 CrossRef CAS PubMed.
- M. Xu, X. Wu, B. Jie, X. Zhang, J. Zhang, Y. Xin and Y. Guo, Cell Biochem. Funct., 2014, 32, 464–469 CrossRef CAS PubMed.
- Y. Yang, W. Wang, Y. Liu, T. Guo, P. Chen, K. Ma and C. Zhou, Dev., Growth Differ., 2012, 54, 557–565 CAS.
- C. Kretz-Remy and A. P. Arrigo, Methods Enzymol., 2002, 348, 200–215 CAS.
- J. Kim, J. S. Kim and E. Park, Food Chem. Toxicol., 2013, 62, 199–204 CrossRef CAS PubMed.
- N. Azad, A. Iyer, V. Vallyathan, L. Wang, V. Castranova, C. Stehlik and Y. Rojanasakul, Ann. N. Y. Acad. Sci., 2010, 1203, 1–6 CrossRef CAS PubMed.
- U. Gregorc, S. Ivanova, M. Thomas, V. Turk, L. Banks and B. Turk, Biol. Chem., 2005, 386, 705–710 CrossRef CAS PubMed.
- L. A. Allan and P. R. Clarke, FEBS J., 2009, 276, 6063–6073 CrossRef CAS PubMed.
- L. Piconi, L. Quagliaro, R. Assaloni, R. Da Ros, A. Maier, G. Zuodar and A. Ceriello, Diabetes/Metab. Res. Rev., 2006, 22, 198–203 CrossRef CAS PubMed.
- R. K. Ambasta, P. Kumar, K. K. Griendling, H. H. Schmidt, R. Busse and R. P. Brandes, J. Biol. Chem., 2004, 279, 45935–45941 CrossRef CAS PubMed.
- W. Ding, L. Yang, M. Zhang and Y. Gu, Biochem. Biophys. Res. Commun., 2012, 418, 451–456 CrossRef CAS PubMed.
- L. Shabala, E. J. Walker, A. Eklund, S. Randall-Demllo, S. Shabala, N. Guven, A. L. Cook and R. D. Eri, Cell Biochem. Funct., 2013, 31, 603–611 CAS.
- K. Suyama, M. Watanabe, K. Sakabe, Y. Okada, D. Matsuyama, M. Kuroiwa and J. Mochida, Neurosci. Lett., 2011, 504, 271–276 CrossRef CAS PubMed.
- F. Ezquer, M. Ezquer, V. Simon, F. Pardo, A. Yanez, D. Carpio and P. Conget, Biol. Blood Marrow Transplant., 2009, 15, 1354–1365 CrossRef CAS PubMed.
- B. A. Miller, N. E. Hoffman, S. Merali, X. Q. Zhang, J. Wang, S. Rajan, S. Shanmughapriya, E. Gao, C. A. Barrero, K. Mallilankaraman, J. Song, T. Gu, I. Hirschler-Laszkiewicz, W. J. Koch, A. M. Feldman, M. Madesh and J. Y. Cheung, J. Biol. Chem., 2014, 289, 7615–7629 CrossRef CAS PubMed.
- Y. Tampo, S. Kotamraju, C. R. Chitambar, S. V. Kalivendi, A. Keszler, J. Joseph and B. Kalyanaraman, Circ. Res., 2003, 92, 56–63 CrossRef CAS PubMed.
- L. Gao, W. Wang, Y. L. Li, H. D. Schultz, D. Liu, K. G. Cornish and I. H. Zucker, Am. J. Physiol.: Heart Circ. Physiol., 2005, 288, H2271–H2279 CrossRef CAS PubMed.
- D. Wang, X. de Deken, M. Milenkovic, Y. Song, I. Pirson, J. E. Dumont and F. Miot, J. Biol. Chem., 2005, 280, 3096–3103 CrossRef CAS PubMed.
- W. Song, J. Pu and B. He, Mol. Med. Rep., 2014, 10, 2764–2770 CAS.
- C. Garrido, L. Galluzzi, M. Brunet, P. E. Puig, C. Didelot and G. Kroemer, Cell Death Differ., 2006, 13, 1423–1433 CrossRef CAS PubMed.
- S. Savitha and C. Panneerselvam, Mech. Ageing Dev., 2006, 127, 349–355 CrossRef CAS PubMed.
- X. Yang, H. Shao, W. Liu, W. Gu, X. Shu, Y. Mo, X. Chen, Q. Zhang and M. Jiang, Toxicol. Lett., 2015, 234, 40–49 CrossRef CAS PubMed.
- J. D. Malhotra and R. J. Kaufman, Antioxid. Redox Signaling, 2007, 9, 2277–2293 CrossRef CAS PubMed.
- J. D. Malhotra and R. J. Kaufman, Semin. Cell Dev. Biol., 2007, 18, 716–731 CrossRef CAS PubMed.
- D. Ron and P. Walter, Nat. Rev. Mol. Cell Biol., 2007, 8, 519–529 CrossRef CAS PubMed.
- S. Oyadomari and M. Mori, Cell Death Differ., 2004, 11, 381–389 CrossRef CAS PubMed.
- R. Schreck, P. Rieber and P. A. Baeuerle, EMBO J., 1991, 10, 2247–2258 CAS.
- E. Maloney, I. R. Sweet, D. M. Hockenbery, M. Pham, N. O. Rizzo, S. Tateya, P. Handa, M. W. Schwartz and F. Kim, Arterioscler., Thromb., Vasc. Biol., 2009, 29, 1370–1375 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |