
An integrated flow and microwave approach to a broad spectrum protein kinase inhibitor†
Abstract
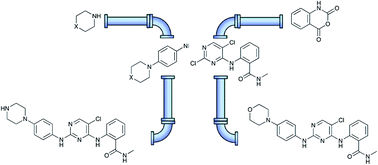
The protein kinase inhibitor CTx-0152960 (6, 2-((5-chloro-2-((4-morpholinophenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide), and the piperazinyl analogue, CTx-0294885 (7, 2-((5-chloro-2-((4-piperazin-1-ylphenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamide), were prepared using a hybrid flow and microwave approach. The use of flow chemistry approaches avoided the need for Boc-protection of piperidine in the key SNAr coupling with 1-fluoro-4-nitrobenzene. Microwave coupling of 4-morphilinoaniline 8 and 4-(piperazine-1-yl)aniline 9 with 2-(2,5-dichloropyrimidine-4-ylamino)-N-methylbenzamide 10, proved to be the most efficacious route to the target analogues 6 and 7. This hybrid methodology reduced the number of synthetic steps, gave enhanced overall yields and increased atom economy through step reduction and minimal requirement for chromatographic purification, relative to the original batch synthesis approach.
 Please wait while we load your content...
Please wait while we load your content...

