
Synthesis of hyperpolarizable biomaterials at molecular level based on pyridinium–chitosan complexes†
Antonio Franconettia,
Lidia Contreras-Bernala,
Rafael Prado-Gotorb and
Francisca Cabrera-Escribano*a
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Sevilla, C/Profesor García González 1, 41012 Sevilla, Spain. E-mail: fcabrera@us.es; Tel: +34 954556868
bDepartamento de Química Física, Facultad de Química, Universidad de Sevilla, C/Profesor García González 1, 41012 Sevilla, Spain
First published on 26th August 2015
Abstract
Potential NLO-phore materials based on chitosan are described for the first time in this study. A series of fluorescent and quaternized pyridinium–chitosan derivatives have been synthesized by reaction of this polymer with easily available tunable pyrylium tetrafluoroborate salts. Among other spectroscopic techniques, 19F NMR, 13C CPMAS NMR and 2D diffusion experiments, were used to confirm the structures of the new pyridinium–chitosan complexes that show high fluorescence intensity. Degrees of N-substitution were achieved lower than 4.3%, allowing the original physicochemical properties of the biopolymer to be preserved. DFT calculations have been performed to investigate the molecular features related to the NLO properties in these compounds. NLO behavior was found to be clearly dependent on the nature and location of the substituent into the pyridinium core. Theoretical data reveal a large permanent dipolar moment, polarizabilities and hyperpolarizabilities making these molecules promising candidates as supramolecular devices exhibiting NLO properties with potentially enhanced solvatochromic properties.
Introduction
Nowadays, the interest in polymer chemistry and their multiple properties makes it a promising area to highlight. Specially, polysaccharides widespread in nature have relevant importance due to eco-friendly features providing a wide working window in numerous areas of everyday life. In this context, chitosan has emerged as a β(1 → 4) linked D-glucosamine polymer with a number of advantages such as non-toxicity, biocompatibility, and biodegradability.1,2 Chitosan has been widely used in multiple areas such as wastewater treatment,3,4 cosmetics5 or food industry6 as well as drugs delivery systems due to its capability of encapsulate different compounds both ionic and covalently bonded.7–9 Moreover, this biopolymer presents a polycationic nature as a consequence of their solubility in acidic media (e.g. acetic acid, hydrochloric acid or lactic acid, among others). For its ability to shape electrostatic interactions, numerous examples for DNA and gene delivery applications10,11 including gene therapy have been reported.12 More recently, all these features have been expressed to nanoscale level developing nanocarriers13,14 including different metallic-chitosan nanoparticles syntheses.15Control of chemical reactivity of the free amino or hydroxyl groups is an essential task to design chitosan derivatives. In this context, typical modifications include N-alkylation by reduction of imine derivatives,16 formation of amides by coupling with carboxylic acids17 and ring-opening oxirane.18 These modifications greatly modulate the biopolymer physical and biological properties. Thus, lightly modified chitosan derivatives such as carboxymethyl19 and quaternized20 chitosans leave water-soluble polymers that are able to increase their applications. Additionally, by using selective reactions, the synthesis of fluorescent chitosan,21 chitosan with remarkable amphiphilic character22 or antibacterial chitosan23 derivatives have been described.
In modern-day, systems exhibiting nonlineal optical (NLO) properties have been developed for multiple applications including optical communication, laser harmonic generation or data storage.24 To generate this kind of materials involving second and third-order optical nonlinearities, such as inorganic salts (e.g. LiNbO3 or SrTiO3),25 organometallic complexes26 and π-extended organic compounds,27–29 different available strategies have been established.
Several requirements at molecular level including large dipole moments (μ), polarizability (α) and first hyperpolarizability (β, also called second-order polarizability) must be considered, as well as visible light transparency and thermal and mechanical stability. In addition, for second order NLO behavior, a non-centrosymmetric disposition of the chromophores is required.30 Supported by theoretical studies, some organic push–pull compounds have an important role as NLO switches due to their capability to exhibit tunable chemical forms provoked by changes on optical and/or electrical properties including pH,31 photo-induced ring-opening or ring-closure,32 isomerizations,33 photochromism34 and, in summary, modified redox/proton transfer features.
The particular structure of pyridinium dyes (e.g. pyridinium N-phenolate and related compounds) with high level of conjugation have been extensively exploited for designing NLO-phore. Furthermore, solvatochromic properties, changes on electronic properties as a function of solvent, low-lying charge-transfer (CT), X-ray structures and DFT calculations for these compounds have been reported.35–39 On the other hand, organic NLO-phores linked to a polymeric backbone such as poly(methylmethacrylate) or phthalocyanines have been recently described.40,41 Despite the fact that chitosan has been used as film to support cyclodextrins42 acting as molecular NLO building blocks, to our knowledge there are no reported works to date on NLO switches anchored to any biopolymeric platform.
In this article, we report a novel type of fluorescent and quaternized pyridinium–chitosan derivatives as donor–π–acceptor (D–π–A) synthesized by an unprecedented reaction for this polymer. A number of techniques as 19F NMR, diffusion experiments or fluorescence measurements were applied to ensure the proposed structures. Theoretical DFT calculations to study physical features related to NLO properties in these compounds have been performed.
Experimental section
Materials
All chemicals were purchased and used without further purification. Low molecular weight chitosan (CS 1) was purchased from Aldrich as well as the high purity chitosan (CS 2), obtained by Agaricus bisporus, with a crystalline structure in α-form. Degree of deacetylation (DD) of 84 and 83%, respectively was determined by 1H NMR. Chitosan from crab skeletons (Procambarus clarkii) (CS 3) showing a DD of 91%, was produced by deproteinization and deacetylation process. Molecular weight of CS 1, CS 2 and CS 3, determined by HPLC/SEC chromatography, were 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 875, 116390 and 69871 g mol−1, respectively. Evaporations were conducted under reduced pressure. TLC was performed on silica gel plates (DC-Alufolien F254, E. Merck); detection of compounds was accomplished with UV light (254 nm) and by charring with ethanolic H2SO4.
875, 116390 and 69871 g mol−1, respectively. Evaporations were conducted under reduced pressure. TLC was performed on silica gel plates (DC-Alufolien F254, E. Merck); detection of compounds was accomplished with UV light (254 nm) and by charring with ethanolic H2SO4.
NMR spectroscopy
1H NMR spectra were acquired on a Bruker Avance III 700 MHz spectrometer at 300 K equipped with a 5 mm CPQCI probehead. A solution 1:0.85 D2O/AcOD (7 mg mL−1) was used to prepare the samples and a solution (0.1 mM) of 3-(trimethylsilyl)-1-propanesulfonic acid sodium salt (DSS sodium salt) was used as internal reference [in our case, δ = 2.91 (t), 1.77 (p), 0.60 (t), 0.00 (s)]. Degree of deacetylation (DD) was determined in 2% DCl solution as previously described by Fernandez-Megia et al.43,44 A pulse of 90° of 8.80 μs with relaxation time (D1) of 10 s and 64 scans were optimized for degree of N-substitution calculations. This degree or extent of N-substitution (DS) was calculated as followed:| DS = (IAr/n)/(IH2–H6/6) |
19F NMR spectra were acquired on a Bruker Avance 500 MHz spectrometer equipped with a 5 mm BBFO probehead incorporating a z-gradient coil. A high-power 90° pulse of 14.8 μs and a recycle delay of 5 s were used as acquisition parameters. The 19F chemical shifts were referenced indirectly to CCl3F (Ξ = 94.094011).45
Yield calculation for chitosan derivatives
Yield (%) calculations of synthesized pyridinium salts were made following the described procedure.47 Nevertheless, a convenient modification of this procedure has been carried out. Thus, mass of chitosan (CS 1) employed was corrected taking into account the relative humidity (9.874%) obtained from thermal analysis using TGA-DSC (Fig. S1†) optimized for a ramp of temperature of 10 °C min−1 and temperature range between 20 °C and 400 °C. Nitrogen gas (100 mL min−1) was used for cooling and purging of the sample compartment.Infrared spectroscopy
Infrared (IR) spectra were recorded on a Jasco FT/IR-4100 spectrophotometer using 30 scan per sample at a resolution of 4 cm−1.Fluorescence emission
Fluorescence measurements were carried out at 298.2 K in a spectrofluorimeter (Hitachi F-2500), interfaced to a PC for the reading and handling of the spectra. A mixture of AcOH/H2O/MeOH was used as solvent. Intensity measurements were performed at compounds concentrations low enough to avoid inner filter effects (and other artifacts). For this reason, in all cases, the sample absorbance measured at the excitation wavelength did not exceed 0.1. The selected excitation wavelength corresponded to the maximum of the UV-vis spectra for each derivative and total luminescence intensity was recorded from 300–700 nm. It was checked that the results were independent of the excitation wavelength, provided that this one was in the range from 300 to 550 nm.Computational details
Density functional theory (DFT) calculations with the Becke 3 Lee–Yang–Park (B3LYP) exchange correlation functional at 6-31G(d,p) level theory were employed by means Gaussian 09.48 During the optimization process no symmetry constraints have been enforced and equilibrium structure was verified from absence of imaginary frequencies. To simplify this calculation, possible conformers thanks to rotation of hydroxymethyl moiety were restricted by using 6-deoxy-D-glucopyranoside as model. Furthermore, a methylation of hydroxy groups at C-1 and C-4 position to simulate the glycosidic bond linked β(1 → 4) was also applied. Physical properties such as dipole moments, polarizability, hyperpolarizability and HOMO–LUMO molecular orbital were computed.Synthesis and characterization data
Pyrylium dyes (1–6) were synthesized according to literature.49 Compound assignations are available in ESI.† Compound 5 was synthesized with modifications of the previously described procedure.General procedure (a). To a solution of CS 1 (100 mg) in aqueous acetic solution (25 mL, pH 4.0–4.6), a solution of pyrylium tetrafluoroborate salt (0.30 mmol) in MeOH (100 mL) was added. The resulting solution was heated to 65 °C until it was noted by TLC (MeOH as eluent) absorption at UV (254 nm) at the origin, corresponding to the polymer that incorporated the aromatic unit. The reaction mixture was left to reach the room temperature and then was treated with 4 M NaOH (pH 11.8–12.5). The resulting solid was filtered off and successively washed with H2O (2 × 2 mL), EtOH (2 × 4 mL), CH2Cl2 (2 × 2 mL) and acetone (2 × 2 mL) to give the corresponding pure chitosan–pyridinium salt.
General procedure (b). To a solution of CS 1 (100 mg) in aqueous acetic solution (25 mL) fitted to pH 6.3, a solution of pyrylium tetrafluoroborate salt (0.30 mmol) in MeOH (100 mL) was added. The resulting solution was heated to 65 °C for 24 h and then AcOH was added until pH value of 5.1. The solution was heated to 65 °C until monitoring the reaction by TLC (MeOH), as above, indicate an incorporation of salts. The reaction mixture was left to reach the room temperature and then was treated with 4 M NaOH (pH 12.4). The resulting solid was filtered off and successively washed with H2O (2 × 2 mL), EtOH (2 × 4 mL), CH2Cl2 (2 × 2 mL) and acetone (2 × 2 mL) to the corresponding pure chitosan–pyridinium salt.
Application of the General procedure (a) for the reactions of CS 2 and CS 3 with 2,4,6-triphenylpyrylium tetrafluoroborate 1, gave compound 13 (61 mg, 90%) and 14 (55 mg, 65%), respectively.
:1) and stirred over 30 min. The solution was alkalinized by addition of 4 M NaOH to pH 12.0. The resulting solid was filtered off and successively washed with H2O (2 × 2 mL), EtOH (2 × 4 mL), CH2Cl2 (2 × 2 mL) and acetone (2 × 2 mL) to give the corresponding pure chitosan–pyridinium compound 10 (60 mg, 70%) as a yellow-pale solid. DS: 0.5%; Rf (MeOH): 0; IR νmax 3326, 2873, 1648, 1579, 1417, 1372, 1313, 1151, 1061, 1025 and 895 cm−1; UV-vis (AcOH/H2O/MeOH): λmax 410 and 270 nm; fluorescence (AcOH/H2O/MeOH): λem 376 nm; 1H NMR (700 MHz, CD3COOD/D2O): δ 7.90–7.37 (21H, m), 4.93 (1H, s), 4.63 (1H, s), 3.95–3.75 (6H, m), 3.24 (1H, s) and 2.05 (3H, m); 13C CPMAS NMR (150.9 MHz): δ 174.3, 142.7, 128.3, 105.3, 83.1, 75.4, 61.2, 57.7 and 23.7; 19F NMR (470 MHz, CD3COOD/D2O): δ −150.03 (10BF4−) and −156.00 (11BF4−).:1) and stirred over 30 min. The solution was alkalinized by addition of 4 M NaOH to pH 12.5. The resulting solid was filtered off and successively washed with H2O (2 × 2 mL), EtOH (2 × 4 mL), CH2Cl2 (2 × 2 mL) and acetone (2 × 2 mL) to give the corresponding pure chitosan–pyridinium compound 12a (63 mg, 73%) as a cream colour solid. DS: 0.4%; Rf (MeOH): 0; IR νmax 3326, 2873, 1648, 1560, 1417, 1372, 1313, 1151, 1061, 1025 and 895 cm−1; UV-vis (AcOH/H2O/MeOH): λmax 419, 359 and 310 nm; fluorescence (AcOH/H2O/MeOH): λem 451 nm; 1H NMR (700 MHz, CD3COOD/D2O): δ 7.89–6.84 (14H, m), 4.92 (1H, s), 4.63 (1H, s), 3.94–3.75 (6H, m), 3.37 (3H, s), 3.24 (1H, s) and 2.05 (3H, m); 13C CPMAS NMR (150.9 MHz): δ 173.3, 141.1, 136.5, 104.5, 82.6, 74.4, 60.1, 56.9, 24.1 and 22.6. 19F NMR (470 MHz, CD3COOD/D2O): δ −102.25 (C–F).Results and discussion
Synthesis and characterization of pyridinium–chitosan dyes
For designing of controlled properties into polymeric backbone, physicochemical features including morphological structure, degree of deacetylation or molecular weight must be understood. Besides, homogeneous modification of chitosan previously requires its dissolution in acidic aqueous media.Chitosan complexes 7–14 were synthesized (Scheme 1) from electronically tunable pyrylium salts taking into account that we focused our attention in slightly modified chitosan amino derivatives searching for the original biopolymer properties are preserved. Indeed, degrees of N-substitution were not higher than 4.3%. Reaction of all the pyrylium dyes with the biopolymer was evidenced (TLC) after 2 days, however the reaction times were delayed by several days (see Experimental section) to ensure a measurable substitution pattern. It is worthy to note that DS parameter proves to be pH-dependent. In particular, for compounds 7 and 8 the lowest pH value (pH 4.3 and 4.2, respectively) gave a DS of 1.4% in the case of 7 and a 1.2% for compound 8 whereas the highest pH value (pH = 5.1) allowed to a DS of 4.3% and 3.4%, respectively.
 | ||
| Scheme 1 Synthesis of pyridinium–chitosan derivatives (7–12). | ||
Compounds 13 and 14 have the same structure at pyridinium moiety that compound 7, however, chitosan backbone is different at morphological level. These compounds, 13 and 14, were synthesized from polymers CS 2 and CS 3, respectively. A comparative analysis of DS values for compounds 7, 13 and 14 gives interesting information. For compound 7, synthesized from CS 1, as indicated above, a DS value of 1.4% was obtained at pH 4.3. For compound 14, obtained from CS 3, a higher DS value (1.6%) results at pH 4.5. On his turn, compound 13 prepared from the high purity chitosan (CS 2), provides at pH 5.0 the lowest DS value, 0.5% (Table 1). These features indicate that DS value is related to the degree of deacetylation (DD) and molecular weight of the starting chitosan. The higher the molecular weight is, the lower the DS value. By the contrary, a higher DD involves a higher number of reactive amino groups on the biopolymer backbone, and a higher resulting DS.
| Compound | R1 | R2 | DS (%) | Yield (%) | λed (nm) |
|---|---|---|---|---|---|
| a Corresponding to compound 13 from CS 2.b Corresponding to compound 14 from CS 3.c Synthesized at pH 5.1.d Excitation wavelengths correspond to the maximum of the UV-visible spectra for each complex. | |||||
| 7 | H | H | 1.4 (0.5a, 1.6b, 4.3c) | 97 (90a, 65b, 94c) | 465 |
| 8 | H | F | 1.2 (3.4c) | 87 (77c) | 354 |
| 9 | Ph | F | 1.1 | 77 | 386 |
| 10 | Ph | H | 0.5 | 70 | 376 |
| 11 | Ph | Me | 1.2 | 99 | 392 |
| 12a | OMe | F | 0.4 | 73 | 451 |
Conventional characterization of starting polymeric material by using Fourier transform (FT-IR) infrared analysis was performed. Commercial chitosan (CS 1) typically shows bands at 3326 cm−1 corresponding to O–H and N–H stretching vibration mode of hydroxyl and amino groups with intermolecular hydrogen bonds. A band at 2873 cm−1 is assigned to C–H stretch of carbohydrate moiety. Moreover, amide I and II bands are observed at 1648 and 1575 cm−1 respectively. Less important bands at 1417, 1372 and 1313 cm−1 for CH2, C–H bending mode and amide III respectively can be assigned. On the other hand, the most interesting bands are the ones corresponding to asymmetric C–O–C stretching for glucosidic linkage at 1151 cm−1, C–O stretching bands for secondary hydroxyl groups at 1061 cm−1 and C–O stretching bands at 1025 cm−1 for primary hydroxyl groups. These bands were applied for evaluating the regioselectivity of these reactions. Thus, absorbance peaks ratio A1025/A1061 must be remained unchanged for N-substitution but not for O-substitution.50 Last band at 894 cm−1 was assigned to glucosidic bond of chitosan backbone.
FT-IR spectra of derivatives (7–12) were analyzed by comparison with FT-IR of CS 1 described above. In compound 7, any appreciable changes except a band at 1664 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of aromatic rings with respect to CS 1 were observed. For compounds 8, 9 and 12 a band corresponding to C–F vibrations should be observed. Unfortunately the predicted wavenumber for this peak is overlapping with C–O stretching for primary and secondary hydroxyl groups and, as a consequence, taking into account the low DS values, this peak was not observed.
C of aromatic rings with respect to CS 1 were observed. For compounds 8, 9 and 12 a band corresponding to C–F vibrations should be observed. Unfortunately the predicted wavenumber for this peak is overlapping with C–O stretching for primary and secondary hydroxyl groups and, as a consequence, taking into account the low DS values, this peak was not observed.
1H NMR for compound 7 shows characteristic peaks at 4.93 and 4.63 ppm for H-1GluN and H-1GluNAc of chitosan, respectively. Integral relationships of these peaks remain unchanged for 7 and the rest of our derivatives as expected for the same DD. The signals between 3.95 – 3.75 ppm were assigned to H-3, H-4, H-5, H-6 and H-6′ carbohydrate protons. On the other hand, signal at 3.24 ppm corresponds to H-2GluN whereas H-2GluNAc was located in 3.95–3.75 ppm range. Finally, the signal at 2.05 ppm, overlapping with deuterated acetic acid solvent peak, was assigned to CH3 groups of non-deacetylated fraction of chitosan. In all synthesized compounds (7–14) no perceptible changes on chemical shifts were observed for these regions. Under these considerations, we focused our interest on aromatic region signals corresponding to pyridinium moiety. A significant observation for all synthesized compounds is a broadening of signals on this region, indicative of assumption of correlation times (τ) similar to polymer on which they have been incorporated. Furthermore, by using 13C CPMAS NMR in solid state, the expected carbons for the aromatic rings were detected. Compound 11 shows in its 13C spectrum a signal at 31.3 ppm assigned to the CH3 group at para position on phenyl rings (rings B) attached to 2 and 6 positions of pyridinium core. Unfortunately, in compounds 12, the methoxy group is not observed due to an overlap with C-2 of chitosan moiety at 56.9 ppm. Carbons C-2′, C-4′ and C-6′ of pyridinium core are significantly deshielded (signal at 141.1 ppm) showing the same trend that pyrylium precursors.
19F NMR spectroscopy was used to confirm the presence of fluorine atom both as substituent and as part of the BF4− anion. Compounds 8, 9 and 12 show broad multiplets (spectra were recorded without 1H decoupling) at −113.25, −101.50 and −102.25 ppm, respectively for fluorine atoms on phenyl rings. BF4− presents completely different chemical shifts. Thus, δF for tetrafluroborate anions is in the range −144.42 (for compound 11) and −156.00 ppm (for compound 10). Two signals were observed in all spectra associated to both isotopes of boron atom (10B and 11B). When compound 12 was dissolved under vigorous stirring in AcOH–H2O (3:1) mixture, anion exchange from tetrafluoroborate to acetate (compound 12a) was induced. After several scans, compound 12a do not exhibits signal for BF4− in 19F spectrum at the mentioned typical range of δF. As expected for this complex, acetate anion was detected at 24.1 ppm in its 13C CPMAS spectrum.
Both FTIR and NMR techniques do not allow the screening in our lower than 5% range of DS, between a covalent linked derivative and an ionic interaction chitosan-small molecule. As pyrylium compounds are positively charged organic molecules, we suggest that non covalent complexes based on electrostatic interactions between chitosan and pyrylium cation are restricted. It has been established that small compounds have diffusion speeds greater than large molecules. However, when they joined to the macromolecule adopt the properties of this one.51 On this base, our hypothesis was corroborated using diffusion experiments such as diffusion filters and 2D DOSY, that show the same relative diffusion coefficient for aromatic region than for chitosan protons (Fig. 1) corroborating a covalent linked structure. It is worthy to note that diffusion filters bring to light the deshielded H-1 protons at 5.50 ppm for glucosamine units who have been attached the pyridinium moieties.
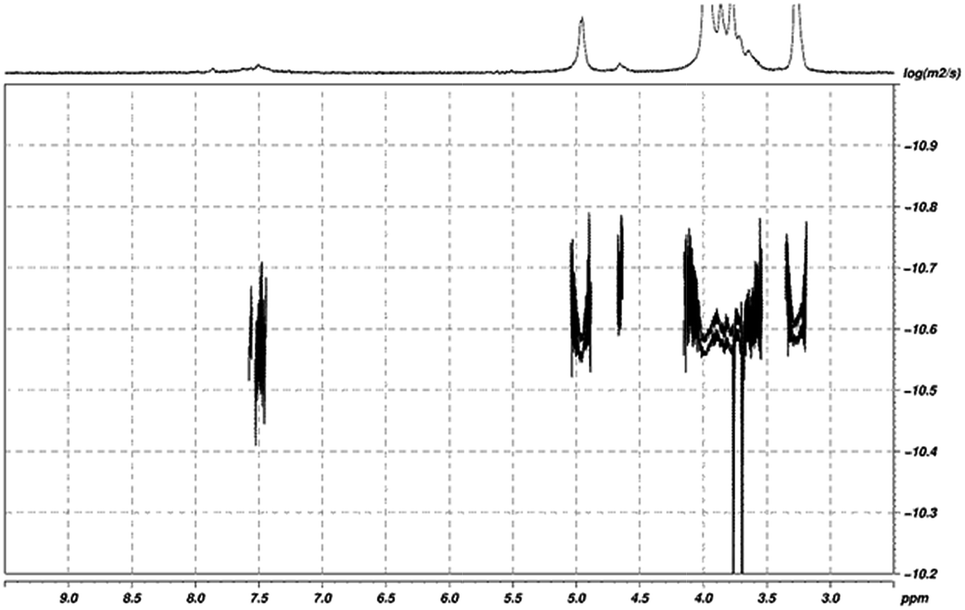 | ||
| Fig. 1 2D DOSY 1H NMR spectrum of compound 7. Region between 8.5 to 7.0 ppm is assigned to aromatic moiety introduced into polymer. | ||
Additionally, the UV-vis and fluorescence characteristics of all the pyrylium49 and pyridinium complexes (1–12) were also studied. The wavelength emission band of the new pyrylium complexes (7–12) was in the range 354–465 nm (Table 1), according to the emission wavelengths showed by 2,4,6-triaryl pyridinium salts previously obtained52,53 from pyrylium salts and more simple amines, such as methyl amine and aniline, as it is can be realized when comparing compound 7 (λem = 465 nm) with 1-methyl-2,4,6-triphenylpyridinium perchlorate52 and 1,2,4,6-tetraphenyl pyridinium tetrafluoroborate53 (λem = 439 nm in ethanol and 480 nm in DMSO/H2O, respectively). Under the conditions applied, pyridinium compounds shown higher relative fluorescence intensity than their pyrylium precursors taking into account low DS (0.4 to 1.4%) targeted. In general, incorporation of substituent groups at the phenyl para positions causes a red-shift in the emission band (Table 1). Furthermore, fluorescence emission wavelength, as expected, is a tunable substituents function. For instance, the fluorescence emission in compound 9 (R1 = Ph; R2 = F) was found to be 386 nm when an excitation wavelength of 309 was employed. On its turn, compound 11 (R1 = Ph; R2 = Me) presents a fluorescence emission peak at 392 nm. As observed, replacing fluorine atoms in the phenyl rings at ortho position of pyridinium moiety for methyl groups provides a displacement in maximum emission to higher wavelength (pair 9/11, Δλe 6 nm). The effect of the substituent on ring A is greater. Thus, in pair 8/9, Δλe = 32 nm by introduction of a phenyl ring and in the pair 8/12 Δλe = 97 nm by introduction of a methoxy group, were measured.
Molecular modeling of poly(pyridinium) complexes
As described in experimental section, DFT calculations were performed to display the conformational behaviors and the theoretical NLO properties of synthesized materials. For this purpose, monomer of chitosan derivatives was chosen which was previously used for this system.54 The model can be applied in our case based on DS value and suggestion of homogeneous distribution of pyridinium moieties. In this system, introducing a fictitious methyl groups (6-deoxy-D-glucopyranoside) instead of hydroxymethyl groups, provokes an inversion on chair conformation of carbohydrate chain. Despite this fact, carbohydrate moiety does not provide in this case a crucial role for the study. Another noteworthy characteristic is the conjugation path that became modified by introducing an aromatic ring at position 4 of the pyridinium cation. Moreover, different substituents with donor or acceptor electronic functions were introduced according with synthesized compounds. In all cases, phenyl ring at para position (ring A) is rotated from pyridinium core with angles of 32.6°, 32.4°, 45.3° and 31.1° for model of compounds 7, 8, 9 and 11, respectively (Table S2† and Fig. 2). Additional phenyl substituent attached to ring A is rotated 37.5° and 37.2° for compounds 9 and 11, respectively.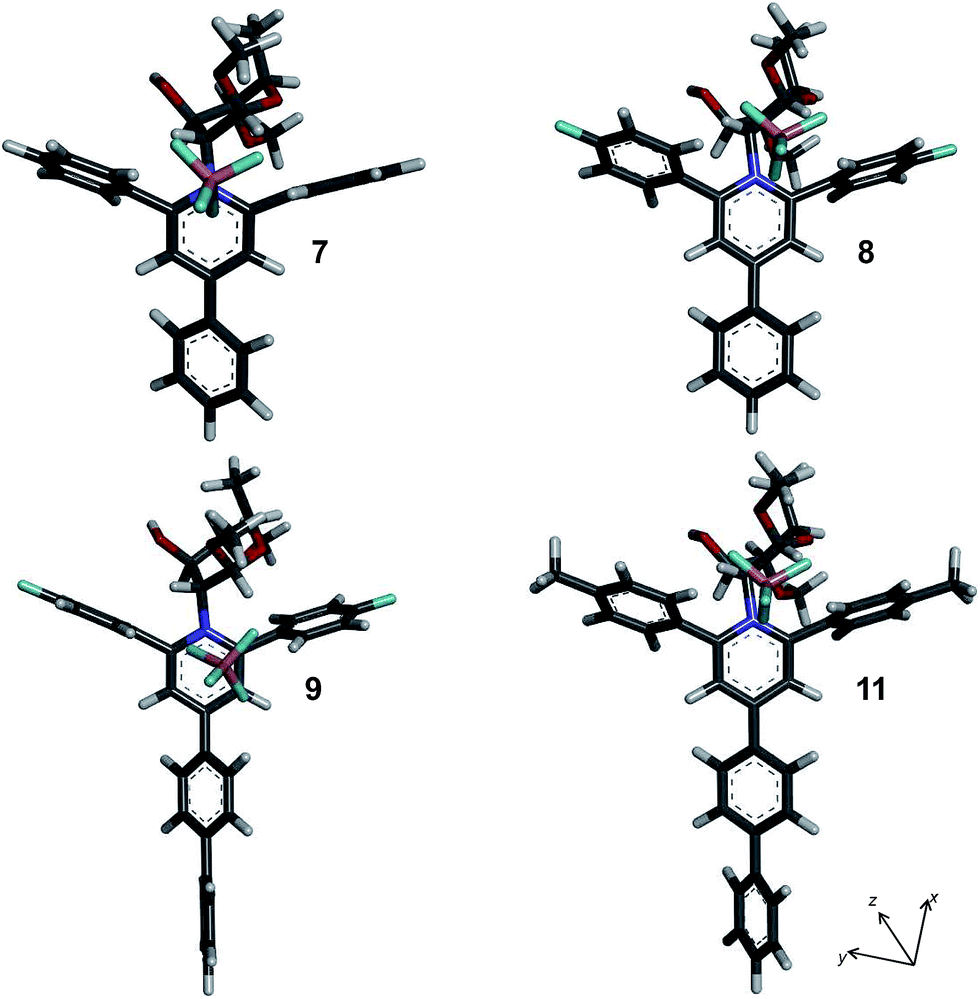 | ||
| Fig. 2 Molecular structures for compounds 7–11 at B3LYP 6-31G(d,p) level of theory. | ||
Anion is positioned over charged nitrogen atom in compound 7 (R1 = H; R2 = H, RB⋯N(1) = 3.777 Å), showing RF⋯N(1) = 2.721 Å. In all complexes, stabilizing CH/anion interactions with an ortho position of phenyl ring at 6 position (range of distance RF⋯H(6) = 2.223–2.125 Å), minimum for compound 8, exist on the basis of the theoretical results.
For monomer of compound 9 (R1 = Ph; R2 = F), an important effect on dipole moment (μ 11.4 D) by movement of BF4− anion over C–5/C–6 bond and its tripod-type interaction with C–H of anomeric carbon, methoxy group and ortho proton on aromatic ring at position 6 is suggested (Fig. S2†). Our results reveal that anion nature has also significant influence. Thus, model for compound 12 indicates that static dipole moment depends of counter-anion for complexes. A major dipole moment calculated for BF4− (μ 16.4 D) was diminished with acetate ion (compound 12a, μ 8.5 D) and drastically decreases without anion (μ 5.8 D). It is relevant that for these compounds binding energy calculated for this stabilizing interaction was −83.8 kcal mol−1 for compound 12 (−99.2 kcal mol−1 for compound 12a).
It is known the optical properties for NLO-phores attached to polymer backbone can highly change from monomer on the basis of aggregation processes which often modify the local interaction geometries of corresponding monomer. In particular, first hyperpolarizability undergoes dramatically changes depending on the aggregate nature.55 However, aggregate formation is not considered in gas phase calculations.
Theoretical results (Table 2) show that weakly donating groups as phenyl ring have opposite effect on molecular parameters. Thus, a low influence on dipole moment in pair 10/7 (Δμ 0.4 D) is observed. Surprisingly, for pair 8/9 containing fluorine atoms on 2,6 positions aromatic rings, an important variation on this parameter is calculated (Δμ 3.3 D). Instead, first-order hyperpolarizability, βtot, drastically increases for compounds with a phenyl substituent on ring A [9.03 (7) to 26.78 (10); and 7.08 (8) to 25.23 (9); p-nitroaniline used as reference: 7.98, in 10−30 esu]. Introducing a strong donating group as OMe provokes great changes on studied dipole moment (16.4 D) and hyperpolarizability (21.70 × 10−30 esu).
| Compound | μx | μy | μz | μg (D) | 〈α〉a | Δαb | βtotc | EHOMO–LUMO (eV) |
|---|---|---|---|---|---|---|---|---|
| a Mean polarizabilities (〈α〉, in a.u.) were calculated from (αxx + αyy + αzz)/3.b Anisotropy of polarizabilities Δα were calculated from ([(αxx − αyy)2 + (αyy − αzz)2 + (αzz − αxx)2]/2)1/2.c First-order hyperpolarizabilities (βtot, in 10−30 esu) were calculated from (βx2 + βy2 + βz2)1/2 obtained at the RHF 6-31G(d,p) level of theory; βi (i = x, y or z) is defined as is described in ref. 56.d Including solvent effect (H2O) by PCM model.e p-Nitroaniline is used as reference at the same level of theory. | ||||||||
| 7 | −5.5 | −2.5 | −12.8 | 14.2 | 367 | 116 | 9.03 | 3.88 |
| 8 | −5.7 | −3.3 | −13.2 | 14.7 | 370 | 129 | 7.08 | 3.79 |
| 9 | 1.2 | 1.7 | −11.2 | 11.4 | 451 | 221 | 25.23 | 3.87 |
| 10 | −6.9 | −2.0 | −12.8 | 14.6 | 458 | 235 | 26.78 | 3.84 |
| 10d | −11.4 | −5.4 | −18.6 | 22.4 | 609 | 274 | 57.83 | 3.67 |
| 11 | 6.9 | 0.9 | −12.7 | 14.5 | 490 | 243 | 26.07 | 3.73 |
| 12 | −9.0 | −1.1 | −13.7 | 16.4 | 440 | 176 | 21.70 | 3.84 |
| 12a | −5.6 | −0.3 | −6.4 | 8.5 | 414 | 149 | 19.64 | 2.49 |
| Ref.e | −7.1 | 0.0 | 0.8 | 7.2 | 86 | 95 | 7.98 | 4.29 |
| Ref.d,e | −10.1 | 0.0 | 0.7 | 10.2 | 124 | 157 | 31.81 | 3.83 |
In general, substituent attached to phenyl rings located at position 2 and 6 of the pyridinium moiety (ring B) have a minimal influence on NLO properties at molecular level. For instance, comparing compounds 9 (R2 = F), 10 (R2 = H) and 11 (R2 = Me) only a Δμ 0.1D for the incorporation of methyl groups on para position at 2,6-diphenyl moieties (pair 10/11) and small changes on βtot are observed. In particular, both methyl and fluorine substituents produce a slightly decrease on βtot value (pair 10/9 Δβ 1.55 and pair 10/11 Δβ 0.71, in 10−30 esu). The studied β values are maximized for x axes in all cases.
Among desirable molecular features for maximized NLO properties a high polarizability (α), as well as a high difference of dipole moment (Δμ) in ground (μg) and excited (μe) state are required. According to substituents on aromatic rings and conjugation path, the polarizability values are in the range 367–609 a.u. (86 and 124 a.u. for the reference compound p-nitroaniline), following the same trends as β values. Maximum value for compound 10 taking into account the water effects was found. In this context, when solvent effects are considered applying the PCM model an increase of dipole moments and hyperpolarizabilities were also observed.
In addition to NLO properties, this kind of compounds provides other desirable qualities. Large permanent dipole moments have an important role as probe for both dipole–dipole and dipole-induced dipole interaction and consequently an enhancement on their solvatochromic properties is expected.57 Furthermore, HOMO–LUMO gap for studied compounds (from 2.49 to 3.88 eV), also implicated in CT processes, must be the lowest as possible due to the high sensitive of β for this parameter.58 The promising compound 10 with potential characteristics as NLO-catiophore shows a HOMO–LUMO gap of 3.67 eV (Table 2 and Fig. S3†).
Conclusions
We reported here a novel type of fluorescent and quaternized pyridinium–chitosan derivatives synthesized by an unprecedented reaction of this bipolymer with tunable pyrylium tetrafluoroborate salts. With the aim to preserve the original physicochemical properties of the biopolymer, degrees of N-substitution not higher than 5% were seeked. Indeed, degrees of N-substitution were lower than 4.3% in all cases.Pyridinium–chitosan derivatives show higher fluorescence intensity in the range 354–465 nm. 19F NMR, 13C CPMAS NMR and diffusion experiments among other spectroscopic techniques were used to confirm the proposed structures. Particularly, diffusion-filtered experiments were effectively applied providing a fast and simple tool to corroborate the attachment to polymer backbone.
Regarding NLO properties, substituent attached to phenyl rings located at position 2 and 6 of the pyridinium moiety (rings B) have a minimal influence. Nevertheless, first-order hyperpolarizability, βtot, drastically increases when an electron donating substituent, phenyl or methoxy, is introduced on ring A. Polarizabilities (α) are notably higher than that of the reference compound and follow the same trends as β values.
In addition to their potential NLO-phore behavior, an enhancement of the solvatochromic properties of described pyridinium–chitosan dyes could be expected as a consequence of the large permanent dipole moments exhibited for this kind of molecules.
Acknowledgements
The authors thank the AECID (Projects A/023577/09 and A/030422/10) and the Junta de Andalucía (FQM 142 and Project P09-AGR-4597) for financial support. The authors also thank the Servicio de Resonancia Magnética Nuclear, CITIUS, Universidad de Sevilla for facilities on NMR experiment performance.Notes and references
- A. Anitha, S. Sowmya, P. T. Sudheesh Kumar, S. Deepthi, K. P. Chennazhi, H. Ehrlich, M. Tsurkan and R. Jayakumar, Prog. Polym. Sci., 2014, 39, 1644 CrossRef CAS PubMed and references therein.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603 CrossRef CAS PubMed and references therein.
- A. Bhatnagar and M. Sillanpää, Adv. Colloid Interface Sci., 2009, 152, 26 CrossRef CAS PubMed.
- E. Repo, L. Malinen, R. Koivula, R. Harjula and M. Sillanpää, J. Hazard. Mater., 2011, 187, 122 CrossRef CAS PubMed.
- L. Calderón, E. Lecumberri, R. Harris, M. Mengibar, N. Acosta and A. Heras, in Marine Cosmeceuticals: Trends and Prospects, ed. S.-K. Kim, CRC Press, Taylor & Francis Group, 2011, ch. 3, p. 39 Search PubMed.
- J. K. Vidanarachchi, M. S. Kurukulasuriya and S.-K. Kim, in Chitin, Chitosan, Oligosaccharides and Their Derivatives: Biological Activities and Applications, ed. S.-K. Kim, CRC Press, Taylor & Francis Group, 2011, ch. 38, p. 543 Search PubMed.
- S. Sanyakamdhorn, D. Agudelo and H.-L. Tajmir-Riahi, Biomacromolecules, 2013, 14, 557 CrossRef CAS PubMed.
- G. Giacalone, A. Bochot, E. Fattal and H. Hillaireau, Biomacromolecules, 2013, 14, 737 CrossRef CAS PubMed.
- K. P. Koutroumanis, K. Avgoustakis and D. Bikiaris, Carbohydr. Polym., 2010, 82, 181 CrossRef CAS PubMed.
- R. Raftery, F. J. O'Brien and S.-A. Cryan, Molecules, 2013, 18, 5611 CrossRef CAS PubMed.
- W. Sajomsang, P. Gonil, U. R. Ruktanonchai, M. Petchsangsai, P. Opanasopit and S. Puttipipatkhachorn, Carbohydr. Polym., 2013, 91, 508 CrossRef CAS PubMed.
- M. Köping-Höggård, K. M. Vårum, M. Issa, S. Danielsen, B. E. Christensen, B. T. Stokke and P. Artursson, Gene Ther., 2004, 11, 1441 CrossRef PubMed.
- For selected review see: M. García-Fuentes and M. J. Alonso, J. Controlled Release, 2012, 161, 496 CrossRef PubMed.
- L. Calderón, R. Harris, M. Cordoba-Diaz, M. Elorza, B. Elorza, J. Lenoir, E. Adriaens, J. P. Remon, A. Heras and D. Cordoba-Diaz, Eur. J. Pharm. Sci., 2013, 48, 216 CrossRef PubMed.
- R. Prado-Gotor, G. López-Pérez, M. J. Martín, F. Cabrera-Escribano and A. Franconetti, J. Inorg. Biochem., 2014, 135, 77 CrossRef CAS PubMed.
- S. Jatunov, A. Franconetti, R. Prado-Gotor, A. Heras, M. Mengíbar and F. Cabrera-Escribano, Carbohydr. Polym., 2015, 123, 288 CrossRef CAS PubMed.
- (a) J. Peréz-Quiñones, R. Szopko, C. Schmidt and C. Peniche-Covas, Carbohydr. Polym., 2011, 84, 858 CrossRef PubMed; (b) P. Peng, X. Cao, F. Peng, J. Bian, F. Xu and R. Sun, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 5201 CrossRef CAS PubMed.
- X. Guan, X. Liu and Z. Su, J. Appl. Polym. Sci., 2007, 104, 3960 CrossRef CAS PubMed.
- L. Zhang, L. Wang, B. Guo and P. X. Ma, Carbohydr. Polym., 2014, 103, 110 CrossRef CAS PubMed.
- (a) H. Tan, S. Guo, S. Yang, X. Xu and T. Tang, Acta Biomater., 2012, 8, 2166 CrossRef CAS PubMed; (b) E. Faizuloev, A. Marova, A. Nikonova, I. Volkova, M. Gorshkova and V. Izumrudov, Carbohydr. Polym., 2012, 89, 1088 CrossRef CAS PubMed.
- Q. Yang, L. Shuai and X. Pan, Biomacromolecules, 2008, 9, 3422 CrossRef CAS PubMed.
- B. Layek, M. K. Haldar, G. Sharma, L. Lipp, S. Mallik and J. Singh, Mol. Pharmaceutics, 2014, 11, 982 CrossRef CAS PubMed.
- N. Vallapa, O. Wiarachai, N. Thongchul, J. Pan, V. Tangpasuthadol, S. Kiatkamjornwong and V. P. Hoven, Carbohydr. Polym., 2011, 83, 868 CrossRef CAS PubMed.
- F. Castet, V. Rodriguez, J.-L. Pozzo, L. Ducasse, A. Plaquet and B. Champagne, Acc. Chem. Res., 2013, 46, 2656 CrossRef CAS PubMed and references therein.
- A. Cetin, R. Kibar, M. Hatipoglu, Y. Karabulut and N. Can, Phys. B, 2010, 405, 2323 CrossRef CAS PubMed.
- For selected review see: K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530 CrossRef CAS PubMed.
- I. Asselberghs, G. Hennrich and K. Clays, J. Phys. Chem. A, 2006, 110, 6271 CrossRef CAS PubMed.
- A. Abbotto, L. Beverina, S. Bradamante, A. Facchetti, C. Klein, G. A. Pagani, M. Redi-Abshiro and R. Wortmann, Chem.–Eur. J., 2003, 9, 1991 CrossRef CAS PubMed.
- Y. Ji, Y. Qian and W. Lu, J. Mater. Chem., 2012, 22, 12375 RSC.
- D. J. Williams, Angew. Chem., Int. Ed. Engl., 1984, 23, 690 CrossRef PubMed.
- E. Bogdan, L. Rougier, L. Ducasse, B. Champagne and F. Castet, J. Phys. Chem. A, 2010, 114, 8474 CrossRef CAS PubMed.
- K. J. Chen, A. D. Laurent and D. Jacquenim, J. Phys. Chem. C, 2014, 118, 4334 CAS.
- D. Lumpi, F. Glöcklhofer, B. Holzer, B. Stöger, C. Hametner, G. A. Reider and J. Fröhlich, Cryst. Growth Des., 2014, 14, 1018 CAS.
- M. Sliwa, S. Létard, I. Malfant, M. Nierlich, P. G. Lacroix, T. Asahi, H. Masuhara, P. Yu and K. Nakatani, Chem. Mater., 2005, 17, 4727 CrossRef CAS.
- Y. Ooyama, K. Kushimoto, Y. Oda, D. Tokita, N. Yamaguchi, S. Inoue, T. Nagano, Y. Harima and J. Ohshita, Tetrahedron, 2012, 68, 8577 CrossRef CAS PubMed.
- H. Langhals, P. Braun, C. Dietl and P. Mayer, Chem.–Eur. J., 2013, 19, 13511 CrossRef CAS PubMed.
- S. Aloïse, Z. Pawlowska, O. Poizat, G. Buntinx, A. Perrier, F. Maurel, K. Ohkawa, A. Kimotoc and J. Abe, Phys. Chem. Chem. Phys., 2014, 16, 1460 RSC.
- J. Abe and Y. Shirai, J. Phys. Chem. B, 1997, 1910 CrossRef CAS.
- S. Roy, S. P. Mondal, S. K. Ray and K. Biradha, Angew. Chem., Int. Ed., 2012, 51, 12012 CrossRef CAS PubMed.
- J. Boixel, V. Guerchais, H. le Bozec, D. Jacquemin, A. Amar, A. Boucekkine, A. Colombo, C. Dragonetti, D. Marinotto, D. Roberto, S. Righetto and R. de Angelis, J. Am. Chem. Soc., 2014, 136, 5367 CrossRef CAS PubMed.
- X. Chen, J. Zhang, W. Wei, Z. Jiang and Y. Zhang, Eur. Polym. J., 2014, 53, 58 CrossRef CAS PubMed.
- P. Fischer, M. Koetse, A. Laschewsky, E. Wischerhoff, L. Jullien, A. Persoons and T. Verbiest, Macromolecules, 2000, 33, 9471 CrossRef CAS.
- E. Fernandez-Megia, R. Novoa-Carballal, E. Quiñoá and R. Riguera, Carbohydr. Polym., 2005, 61, 155 CrossRef CAS PubMed.
- Chitosan spectra for DD calculation were referenced from DSS and Acquisition time (AQ) was modified respect to described by Fernandez-Megia from 1 s to 2 s. The use of NMR tube of 3 mm was not significant for these calculations. DD values were determined from relative peak areas of CH3 and combined H3-H6 and H-2 proton regions disregarding any possible invisible fraction into chitosan. For this novel characteristic see: R. Novoa-Carballal, R. Riguera and E. Fernandez-Megia, Mol. Pharmaceutics, 2013, 10, 3225 CrossRef CAS PubMed.
- (a) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795 CrossRef CAS; (b) R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow, P. Granger, R. E. Hoffman and K. W. Zilm, Pure Appl. Chem., 2008, 80, 59 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260 CrossRef CAS.
- W. Sajomsang, S. Tantayanon, V. Tangpasuthadol, M. Thatte and W. H. Daly, Int. J. Biol. Macromol., 2008, 43, 79 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- A. Franconetti, L. Contreras-Bernal, S. Jatunov, M. Gómez-Guillén, M. Angulo, R. Prado-Gotor and F. Cabrera-Escribano, Phys. Chem. Chem. Phys., 2014, 16, 18442 RSC.
- K. P. Koutroumanis, K. Avgoustakis and D. Bikiaris, Carbohydr. Polym., 2010, 82, 181 CrossRef CAS PubMed.
- J. Jiménez-Barbero, An. Quim., 2003, 2, 115 Search PubMed.
- M. I. Knyazhansky, V. A. Kharlanov and Y. R. Tymiansky, J. Photochem. Photobiol., A, 1998, 118, 151 CrossRef CAS.
- Z. Wang, Y. Fang, J. Sun, A. Qin and B. Z. Tang, Sci. China: Chem., 2013, 56, 1187 CrossRef CAS.
- X. Yang, C. Zhang, C. Qiao, X. Mu, T. Li, J. Xu, L. Shei and D. Zhang, Carbohydr. Polym., 2015, 130, 325 CrossRef CAS PubMed.
- P. C. Ray, P. Bonifassi and J. Leszczynski, J. Phys. Chem. A, 2006, 110, 8963 CrossRef CAS PubMed.
- D. R. Kanis, M. A. Ratner and T. J. Marks, Chem. Rev., 1994, 94, 195 CrossRef CAS.
- V. G. Machado, R. I. Stock and C. Reichardt, Chem. Rev., 2014, 114, 10429 CrossRef CAS PubMed.
- First-order hyperpolarizability increases on the lowering the HOMO–LUMO gap, following the equation: β ∝ μeg2Δμ/Eeg2 where μeg is the transition moment between ground and excited states; Δμ is the dipole moment difference between ground and excited states, and Eeg is the HOMO–LUMO transition.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and spectral data of pyrylium salts, theoretical calculations by B3LYP/6-31G(d,p) method of distances and dihedral angles and cartesian coordinates and energies for pyridinium complexes, molecular orbital and energies for HOMO–LUMO of NLO-phore 10, and 1H, 19F, diffusion experiment and 13C CPMAS NMR spectra of new compounds synthesized. See DOI: 10.1039/c5ra09397j |
| This journal is © The Royal Society of Chemistry 2015 |