
Fabrication and properties of a supramolecular hybrid hydrogel doped with CdTe quantum dots†
Abstract
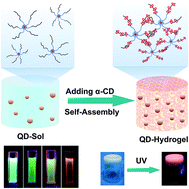
Fluorescent hydrogels incorporated with photoluminescent nanomaterials have achieved promising progress in bio-sensing or bio-labeling, yet the current techniques mostly rely on chemically cross-linking the hydrogel which involves undesirable chemical reactions that may easily disturb the incorporated nanomaterials. In this work, the fabrication of a fluorescent supramolecular hydrogel doped with semiconductor CdTe quantum dots (QDs) is demonstrated. The colloidal QDs were stabilized with synthetic amphiphilic block copolymer, mercaptan-ended poly(ethylene glycol)–poly(ε-caprolactone). The stability and fluorescent properties of the resultant colloidal QDs were evaluated. A fluorescent supramolecular hydrogel was fabricated based on the host–guest self-assembly between the amphiphilic block copolymer on the QD surface and added cyclic oligosaccharide host molecule, α-cyclodextrin (α-CD). The resultant photoluminescent hydrogel was characterized with rheology and X-ray diffraction, as well as photoluminescence spectra measurements. The gelation kinetics and mechanical strength of the supramolecular hydrogel can be modulated by changing the amount of the amphiphilic block copolymer, α-CD, used or the incorporated QDs. These results suggest new opportunities for developing biocompatible optical materials with tunable fluorescent properties and mechanical properties.
 Please wait while we load your content...
Please wait while we load your content...

