
Facile synthesis of glycerol carbonate via glycerolysis of urea catalysed by silicotungstates impregnated into MCM-41
Nilesh Narkhede and
Anjali Patel*
Polyoxometalate and Catalysis Laboratory, Department of Chemistry, Faculty of Science, The M. S. University of Baroda, Vadodara, 390002, India. E-mail: aupatel_chem@yahoo.com; Tel: +91-265-2795552
First published on 9th June 2015
Abstract
The present contribution includes the solvent free environmentally benign route towards synthesis of glycerol carbonate via glycerolysis of urea. The parent as well as mono lacunary silicotungstates impregnated into MCM-41 were used as efficient catalysts. The effect of different reaction parameters on the conversion of glycerol was investigated in order to optimize the reaction parameters for maximum conversion. The activities of both the catalysts have been correlated with the structural features and acidity of the catalysts and the possible mechanism has also been proposed. The catalysts were also recycled up to four times without significant loss in the activity.
1. Introduction
Glycerol is a versatile raw material and produced in large quantities as a by-product from the synthesis of biodiesel. The utilisation of glycerol as a platform chemical represents an opportunity to obtain value-added products from a highly functionalised and inexpensive raw material, and much research has recently been dedicated to finding new chemical pathways for this feedstock.1–3Among the desired products, glycerol carbonate has excellent properties such as low toxicity, good biodegradability and a high boiling point which make it a very attractive chemical for a variety of applications such as a high boiling polar solvent, an intermediate in organic synthesis4 and in the synthesis of polycarbonates,5 polyurethanes6 and in cosmetic and medical institutes as they have low toxicity, volatility, combustibility and good moisturizing ability.7 Besides, they also play a major role as a component of surfactants, paints, coatings and gas-separation membranes.8 Additionally, there is a general consensus that the potential of glycerol carbonate as an anti-explosive additive for gasoline and diesel will result in an unprecedented growth in the coming years.9–11
The traditional and most effective route for the synthesis of glycerol carbonate (GlyC) is the transesterification of glycerol with acyclic organic carbonates (dimethyl carbonate or diethyl carbonate).12,13 However, the carbonates utilised during the transesterification are also typically generated via phosgene utilisation which suffers from the drawback of being a dangerous and environmentally unfriendly reactant or energy intensive routes employing epoxides. The direct reaction of glycerol with CO2 appears very attractive, but it has serious thermodynamic limitations.14 One of the practical routes for carbonylation of glycerol is the use of urea as a carbonate source.11 The major advantage of this method over other processes is that urea is readily available and cheap. Literature survey shows that catalysts with Lewis acid sites such as ZnO,15 Co3O4/ZnO,16 ZnCl2,17 γ-zirconium phosphate,8 HTc–Zn derived from hydrotalcite,13 Sm-exchanged heteropoly tungstate,18 gold supported ZSM-5,11 manganese sulfate11 and metal oxides19 produce high glycerol carbonate yields. To the best of our knowledge, no report in the literature is available for the synthesis of GlyC using silicotungstates despite the fact that they act as highly active solid acid catalysts in many acid catalyzed transformations.20–24 Hence, it was thought of interest to study the application of supported parent as well as lacunary silicotungstates for the synthesis of GlyC and see the effect of lacuna on the catalytic activity of lacunary silicotungstates.
We have successfully established the synthesis of parent (SiW12) as well as mono lacunary silicotungstate (SiW11) impregnated to MCM-41 and characterized by different physicochemical techniques.25,26 In continuation of our previous efforts to explore wider applicability of these catalysts for acid catalysed organic transformations, for the first time, we report synthesis of GlyC over SiW12 as well as SiW11 impregnated to MCM-41. The effect of different reaction parameters was studied for maximum conversion. Based on the results the catalyst activity has been correlated with the structural features and acidity of the catalysts. Further possible mechanism has also been proposed. The catalysts were also verified successfully for recyclability up to four cycles.
2. Experimental
2.1 Materials
All materials used were of A.R. (analytical) grade. 12-Tungstosilicic acid, benzaldehyde, tetraethylorthosilicate (TEOS), glycerol, sodium tungstate, sodium silicate, acetonitrile, n-butylamine and acetone were purchased from Merck.2.2 Synthesis of the catalysts (SiW12/SiW11 impregnated to MCM-41)
MCM-41 was prepared using previously reported procedure.25 1 g surfactant (CTAB) was added to the dilute solution of NaOH (2 M, 3.5 mL NaOH in 480 mL double distilled water) with stirring at room temperature. After the solution became clear, 5 mL TEOS was added drop wise and the gel was aged for 2 h at 60 °C. The resulting material was filtered, washed with distilled water, dried in oven and calcined in air at 550 °C for 5 h.SiW11 was synthesized from individual salts; sodium tungstate and sodium silicate by following the previously reported method.25 Sodium tungstate (0.22 mol, 7.2 g) and sodium silicate (0.02 mol, 0.56 g) were dissolved in 150 mL double distilled water at 80 °C. The pH was then adjusted to 4.8 by dilute nitric acid. The volume of the mixture was reduced to half and the resulting solution was filtered to remove unreacted silicates. The lacunary heteropoly anion was separated by liquid–liquid extraction with acetone. The extraction was repeated until the acetone extract showed the absence of nitrate ions. The extracted sodium salt of mono lacunary silicotungstate was dried at room temperature in air. The resulting material was designated as SiW11 (Na = 6%; W = 63.8%; Si = 0.89%).
A series of catalysts containing 10–40% of SiW12 impregnated to MCM-41 were synthesized by impregnating an aqueous solution of SiW12 (0.1/10–0.4/40 g mL−1 of double distilled water) with MCM-41 (1 g) and dried at 100 °C for 10 h. The resulting materials were designated as 10% SiW12/MCM-41, 20% SiW12/MCM-41, 30% SiW12/MCM-41 and 40% SiW12/MCM-41, respectively.
Similarly, catalysts containing 10–40% of SiW11 impregnated to MCM-41 were synthesized by impregnation method. MCM-41 (1 g) was impregnated with an aqueous solution of SiW11 (0.1/10–0.4/40 g mL−1 of double distilled water) and dried at 100 °C for 10 h. The obtained materials were treated with 0.1 N HCl, filtered, washed with double distilled water and dried at 100 °C in order to convert the Na form of the catalyst in to the proton form. The resulting materials were designated as 10% SiW11/MCM-41, 20% SiW11/MCM-41, 30% SiW11/MCM-41 and 40% SiW11/MCM-41, respectively.
2.3 Characterization techniques
A detailed study on the characterization of synthesized materials can be found in our earlier publications.25,26 The main characterization of the catalysts such as BET, FT-IR, XRD, 29Si-MAS-NMR, and n-butyl amine acidity are given for readers' convenience. The BET surface area measurements were performed in a Micromeritics ASAP 2010 volumetric static adsorption instrument with N2 adsorption at 77 K. Firstly, the samples were degassed under vacuum for 4 h at 150 °C. The pore size distributions were calculated by BJH adsorption–desorption method. For FT-IR spectra, samples pressed with dried KBr into discs were recorded in the wavenumber range of 4000–400 cm−1 by using a Perkin-Elmer spectrometer with four scans and resolution of 2 cm−1. The X-ray powder diffraction (XRD) patterns of the support and the catalyst were measured using a Philips X'Pert MPD system in the 2θ range of 0–60° using CuKα radiation (λ = 1.54056 Å) with scan rate of 0.02° s−1. The 29Si nuclear magnetic resonance (NMR) spectra were recorded on Varian, Mercury plus 300 MHz NMR spectrometer at 121.49 MHz using a 7 mm rotor probe with tetra methyl silane as an external standard. The spinning rate was 5–7 kHz (number of scans-1024).2.4 Determination of acidic strength (potentiometric titration method)
A small quantity (0.1 mL) of 0.05 N, n-butylamine in acetonitrile was added to a suspension of 0.5 g of the catalyst in 50 mL of acetonitrile and the system was stirred at 25 °C. Then, the suspension was potentiometrically titrated against 0.05 N, n-butylamine in acetonitrile. The electrode potential variation was measured with a digital pH meter.The acidity of the catalyst measured by this technique allows us to evaluate the total number of acid sites as well as their acidic strength. In order to interpret the results, it is suggested that the initial electrode potential (Ei) indicates the maximum acid strength of the surface sites and the range where the plateau is reached (meq per g of solid) indicates the total number of acid sites.27 The acidic strength of surface sites can be assigned according to the following ranges: very strong site, Ei > 100 mV; strong site, 0 < Ei < 100 mV; weak site, −100 < Ei < 0 mV and very weak site, Ei < −100 mV.
2.5 Reaction procedure
In a typical carboxylation reaction, glycerol (10 mmol) and urea (10 mmol) were placed in a 50 mL round bottom flask and the catalyst (100 mg) was added. The reaction was performed at 140 °C and N2 was purged in the reaction mixture in order to remove evolved NH3 during the reaction. After completion of the reaction, methanol was added to the reaction mixture and the catalyst was separated by centrifugation. The products were analysed by using Shimadzu 2014 Gas Chromatography (GC) instrument equipped with RTX-5 capillary column (internal diameter: 0.25 mm, length: 30 m). The products were identified by comparison with the standard samples. Products were identified by FT-IR and NMR analysis. The conversion as well as selectivity was calculated on the basis of mole percent of glycerol using the following equations,

2.6 Leaching test
A leaching of the active species from the support makes the catalyst unattractive, and hence, it is necessary to study the leaching of SiW12/SiW11 from the support. Leaching test (ascorbic acid test) was carried out with alcohols and the filtrate of the reaction mixture after completion of reaction in order to check the presence of any leached species according to the method described in the literature.25 In the present study one gram of catalyst with 10 mL of conductivity water was refluxed for 24 h. Then, 1 mL of the supernatant solution was treated with 10% ascorbic acid. Development of blue colour was not observed, indicating that there was no leaching.3. Results and discussion
3.1 Catalyst characterization
The elemental analyses performed on 30% SiW12/MCM-41 (theoretical: W = 19%; Si = 27%; observed: W = 17.9%; Si = 27%) and 30% SiW11/MCM-41 (theoretical: W = 15%; Si = 28%; observed: W = 15.2%; Si = 27.6%) were consistent with theoretical expected values.The values of BET surface area analysis for all the catalysts are presented in the Table 1. It is seen from the Table 1 that surface area, pore diameter as well as pore volume decreased drastically for the catalysts as compared to MCM-41. The overall decrease in surface area of both the catalysts with respect to the support gives the first indication of a chemical interaction between SiW11/SiW12 and MCM-41. It also confirms that active phase is located quite inside the channels of the mesoporous support. It is interesting to note down that the value of surface area and pore diameter of 30% SiW11/MCM-41 is higher than that of 30% SiW12/MCM-41. This may be due to the fact that the removal of W–O unit from the parent SiW12 results in decrease in the size of SiW11 species leading to increase in the available space inside the channels of the support.
| Material | Surface area (m2 g−1) | Pore diameter (nm) | Pore volume (cm3 g−1) |
|---|---|---|---|
| MCM-41 | 659 | 4.79 | 0.79 |
| 30% SiW12/MCM-41 | 349 | 2.92 | 0.26 |
| 30% SiW11/MCM-41 | 536 | 3.96 | 0.63 |
TGA of MCM-41 shows initial weight loss of 6.14% at 100 °C due to the adsorbed water molecules.25 The last 5.92% weight loss above 450 °C may be due to the condensation of silanol groups to form siloxane bonds. After that, the absence of any weight loss shows that support is stable up to 500 °C. The TGA of SiW11 shows the preliminary weight loss of 7% from 30–130 °C due to the removal of adsorbed water molecules.25 Second weight loss of 2.5% at 230 °C may be due to loss of water of crystallization. The steady weight loss after 330 °C indicates the decomposition of SiW11 species. The TGA of 30% SiW11/MCM-41 shows initial weight of 6% up to 150 °C due to the removal of adsorbed water molecules.25 Second weight loss of 2% has been observed up to 250 °C which is due to loss of water of crystallization. No notable loss up to 400 °C indicates the stability of the catalyst up to 400 °C. The TGA of 30% SiW12/MCM-41 show initial weight loss of 4–6% due to the loss of adsorbed water.28 Second weight loss of 2–3% between 150 and 250 °C corresponds to the loss of water of crystallization of Keggin ion. After that another gradual weight loss was also observed from 250 to 500 °C due to the difficulty in removal of water contained in SiW12 molecules inside the channels of MCM-41. Such type of inclusion causes the stabilization of SiW12 molecules inside the channels of MCM-41.
The FT-IR of MCM-41 (Fig. 1a) shows a broad band around 1300 and 1000 cm−1, matching to asymmetric stretching of Si–O–Si. The bands at 460 and 808 cm−1 are attributed to the bending vibration of the Si–O–Si bonds and free silica. The band at 966 cm−1 corresponds to symmetric stretching vibration of Si–OH. The broad absorption band around 3448 cm−1 is the absorption of Si–OH on surface, which offers opportunities for forming the hydrogen bond. The FT-IR spectra of SiW11 (Fig. 1c) shows bands at 987 cm−1 (W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Od), 948 cm−1 (Si–Oa), 886 and 795 cm−1 (W–Ob–W) and 727 cm−1 (W–Oc–W). These bands are in good agreement with the reported one.29 The FT-IR spectra of 30% SiW11/MCM-41 (Fig. 1b) shows bands at 960 cm−1 and 900 cm−1 corresponding to the symmetric stretching of WOd and Si–Oa bonds of SiW11, respectively. Similarly The FT-IR spectrum of 30% SiW12/MCM-41 (Fig. 1e) showed the retention of typical bands for SiW12, at 979 cm−1 and 923 cm−1 corresponding to WOd and Si–Oa symmetric stretching, respectively. The presence of these bands confirms that structure of SiW12/SiW11 is intact even after impregnation to the support. The substantial shift in the bands indicates interaction between SiW12/SiW11 and surface silanol groups of MCM-41.
Od), 948 cm−1 (Si–Oa), 886 and 795 cm−1 (W–Ob–W) and 727 cm−1 (W–Oc–W). These bands are in good agreement with the reported one.29 The FT-IR spectra of 30% SiW11/MCM-41 (Fig. 1b) shows bands at 960 cm−1 and 900 cm−1 corresponding to the symmetric stretching of WOd and Si–Oa bonds of SiW11, respectively. Similarly The FT-IR spectrum of 30% SiW12/MCM-41 (Fig. 1e) showed the retention of typical bands for SiW12, at 979 cm−1 and 923 cm−1 corresponding to WOd and Si–Oa symmetric stretching, respectively. The presence of these bands confirms that structure of SiW12/SiW11 is intact even after impregnation to the support. The substantial shift in the bands indicates interaction between SiW12/SiW11 and surface silanol groups of MCM-41.
 | ||
| Fig. 1 FT-IR spectra of (a) MCM-41, (b) 30% SiW11/MCM-41, (c) SiW11, (d) SiW12 and (e) 30% SiW12/MCM-41. | ||
XRD patterns of MCM-41, 30% SiW12/MCM-41 and 30% SiW11/MCM-41 are shown in Fig. 2. The XRD pattern of the MCM-41 shows a sharp reflection around 2θ = 2° corresponding to (100) plane indicating well-ordered hexagonal structure of MCM-41. The comparison of the XRD patterns of MCM-41 and the catalysts reveals that the mesoporous structure of MCM-41 is rather intact even after impregnation of SiW12/SiW11 species. Further the absence of characteristic peaks of crystalline phase of SiW12 as well as SiW11 in the respective catalysts indicates that the active species are highly dispersed inside the hexagonal channels of MCM-41.
 | ||
| Fig. 2 XRD patterns of (a) MCM-41, (b) 30% SiW11/MCM-41 and (c) 30% SiW12/MCM-41. | ||
The 29Si MAS NMR is the most important method to study chemical environment around the silicon nuclei in mesoporous silica materials. Fig. 3 shows the 29Si MAS NMR spectra of MCM-41, 30% SiW12/MCM-41 and 30% SiW11/MCM-41. The presence of resonance originated from Q2 Si(OSi)2(OX)2, Q3 Si(OSi)3(OH) and Q4 Si(OSi)4 in the catalysts indicates that MCM-41 retains its structure in both the catalysts (Table 2). The spectra of the catalysts are relatively broad and low in intensity as compared to MCM-41. This is due to the strong hydrogen bonding between SiW12/SiW11 and Q2 Si(OSi)2(OH)2 (surface silanol groups) of MCM-41.
 | ||
| Fig. 3 29Si-MAS NMR spectra of (a) MCM-41, (b) 30% SiW12/MCM-41 and (c) 30% SiW11/MCM-41. | ||
| Material | Q2, ppm | Q3, ppm | Q4, ppm |
|---|---|---|---|
| 30% SiW12/MCM-41 | −93 | −103 | −110 |
| 30% SiW11/MCM-41 | −102 | −104.6 | −110.5 |
The plots of the electrode potential as a function of meq amine per g of the catalysts are shown in Fig. 4. It is observed that, both the catalysts contain very strong acid sites. The strength of acidic sites in terms of initial electrode potential is shown in Table 3. It is clear from the Table 3 that the incorporation of SiW12/SiW11 increases the strength of the acid sites of catalysts to a great extent. It is also interesting to note that almost all values are similar in both the catalysts except the acidic strength. The acidic strength of 30% SiW11/MCM-41 is lower than that of 30% SiW12/MCM-41. The reason being, the acidic character of polyoxometalates is mainly due to the acidic addenda atoms i.e. tungsten in the present case and removal of one tungsten–oxygen unit from the parent SiW12 is expected to decrease the acidity of the SiW11. The obtained value is in good agreement with the expected one.
 | ||
| Fig. 4 Potentiometric titration curves of (a) MCM-41, (b) 30% SiW12/MCM-41 and (c) 30% SiW11/MCM-41. | ||
3.2 Catalytic reaction
This study is focused on catalytic activity of both 30% SiW11/MCM-41 as well as 30% SiW12/MCM-41 for solvent free carboxylation of glycerol (Gly) with urea (Scheme 1).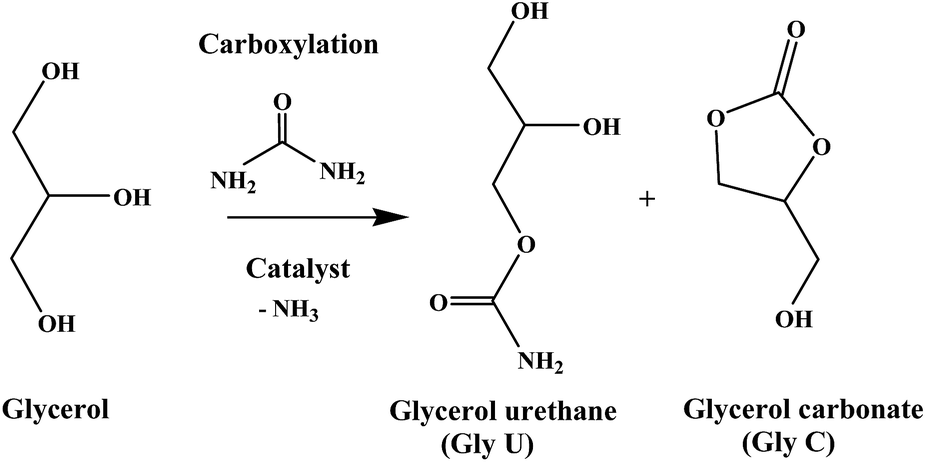 | ||
| Scheme 1 Glycerol carbonate synthesis scheme from glycerol and urea. | ||
3.3 Carboxylation of glycerol over 30% SiW11/MCM-41
The activity of synthesized catalyst, 30% SiW11/MCM-41 was evaluated for the synthesis of glycerol carbonate by glycerolysis of urea. The effect of different reaction parameters on the conversion as well as selectivity for Gly C was evaluated.The effect of %loading of SiW11 on the conversion of glycerol was studied by varying the loading from 10–40% (Fig. 5). It is clear that the increase in the loading of SiW11 linearly increases the conversion of glycerol. This can be correlated with the increase in the catalytically active acidic sites. The selectivity of GlyC was not affected much by the increase in the %loading. The optimum conversion of 55% with 77% selectivity of GlyC was achieved with 30% loading and further increase in the loading does not influence the conversion. Hence, 30% SiW11/MCM-41 was selected for the further studies.
 | ||
| Fig. 5 Effect of %loading on carboxylation of glycerol. Reaction conditions: mole ratio Gly/urea: 1/1; time: 8 h; temperature: 140 °C; catalyst amount: 100 mg. | ||
Fig. 6 shows the influence of glycerol to urea mole ratio on the conversion of glycerol and selectivity of the glycerol carbonate. It is can be noted from the Fig. 6 that with G![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :U molar ratio of 1:1, the conversion of glycerol was 55% with 77% selectivity towards glycerol carbonate. However further increase in the mole ratio did not increase the conversion of the glycerol. It was interesting to note that with increase in the mole ratio selectivity of GlyC was decreased. This is due to the fact that at higher concentration of urea, there is a possibility of formation of by-product by reaction of free hydroxyl group of glycerol with excess urea. As a result 1:1 ratio was considered as optimum for maximum conversion and selectivity of GlyC.
:U molar ratio of 1:1, the conversion of glycerol was 55% with 77% selectivity towards glycerol carbonate. However further increase in the mole ratio did not increase the conversion of the glycerol. It was interesting to note that with increase in the mole ratio selectivity of GlyC was decreased. This is due to the fact that at higher concentration of urea, there is a possibility of formation of by-product by reaction of free hydroxyl group of glycerol with excess urea. As a result 1:1 ratio was considered as optimum for maximum conversion and selectivity of GlyC.
 | ||
| Fig. 6 Effect of Gly:urea mole ratio on carboxylation of glycerol. Reaction conditions: time: 8 h; temperature: 140 °C; catalyst amount: 100 mg. | ||
The studies of effect of catalyst amount on the conversion (Fig. 7) of glycerol suggest that with increase in catalyst amount the conversion of glycerol also increases linearly. However, beyond 100 mg amount of catalyst, the selectivity of GlyC was decreased. The excess catalyst might be favouring the reaction between the product GlyC and urea to yield, 5-hydroxymethyloxazolidine-2-one. Hence, 100 mg of catalyst amount was considered to be optimum.
 | ||
| Fig. 7 Effect of catalyst amount on carboxylation of glycerol. Reaction conditions: mole ratio G/U: 1/1; time: 8 h; temperature: 140 °C. | ||
The effect of reaction time on the conversion of glycerol (Fig. 8) suggests that increase in the reaction time increases the conversion of the glycerol as well as selectivity of GlyC. After 8 h of the reaction time 55% conversion with 77% selectivity of GlyC was observed. On further increase in the reaction time, the coke formation starts.
 | ||
| Fig. 8 Effect of reaction time on carboxylation of glycerol. Reaction conditions: mole ratio G/U: 1/1; temperature: 140 °C; catalyst amount: 100 mg. | ||
The temperature variation study was carried out by varying the temperature in the range of 100 °C to 150 °C (Fig. 9). Maximum conversion was achieved at 150 °C which was considered to be optimum conversion of 62% and selectivity of 75% for GlyC.
 | ||
| Fig. 9 Effect of temperature on carboxylation of glycerol. Reaction conditions: mole ratio G/U: 1/1; time: 8 h; catalyst amount: 100 mg. | ||
The optimized conditions (maximum conversion; 62%, and selectivity; 75% for 30% SiW11/MCM-41) are, mole ratio Gly/urea: 1; temperature: 150 °C; catalyst amount: 100 mg; reaction time: 8 h. Similarly, optimization study for 30% SiW12/MCM-41 was carried out and the final optimized conditions (maximum conversion; 75%, and selectivity; 77% for 30% SiW12/MCM-41) are, mole ratio Gly/urea: 1; temperature: 150 °C; catalyst amount: 100 mg; reaction time: 8 h.
3.4 Control experiments
The carboxylation of glycerol was carried out without catalyst as well as using support, and active species (Table 4). The support was not much active towards the carboxylation and activities of active species are quite comparable with those of the catalysts. Thus, we were successful in heterogenizing SiW12/SiW11 on to the mesoporous support without losing its catalytic activity. Despite the fact that surface area of 30% SiW11/MCM-41 is higher as compared to 30% SiW12/MCM-41 (Table 1), lower conversion was observed for 30% SiW11/MCM-41. The results suggest that activity of the present catalysts does not depend on the surface area of the catalyst. However the activity trends are quite consistent with the strength of the acidic sites (Fig. 4) of both the catalyst. Hence it can be concluded that the present catalytic system is not a surface type heterogeneous in which the catalytic activity is directly proportional to surface area but it is a pseudo-liquid type (I) heterogeneous catalyst where activity is directly proportional to the acidity of the catalyst.30 Further it was observed that catalytic activity of both the catalysts was limited to 62% for 30% SiW11/MCM-41 and 75% for 30% SiW12/MCM-41. This may be due to the fact that liberated NH3 forms ammonium salt with the silicotungstates and thereby poisoning the catalyst even after purging the reaction mixture with N2 in order to remove the excess of NH3 formed.| Catalyst | % Conversion | % Selectivityc | TON |
|---|---|---|---|
| a Reaction conditions: mole ratio Gly/urea: 1/1; time: 8 h; temperature: 150 °C; catalyst amount: a23 mg/b100 mg. TON was calculated from the formula, TON = moles of product/moles of catalyst. cGlycerol carbonate selectivity. | |||
| No catalyst | 19 | 36 | — |
| MCM-41b | 24 | 45 | — |
| SiW11a | 65 | 70 | 820 |
| 30% SiW11/MCM-41b | 62 | 75 | 782 |
| SiW12a | 71 | 75 | 889 |
| 30% SiW12/MCM-41b | 75 | 77 | 939 |
3.5 Heterogeneity test
For the rigorous proof of heterogeneity, a test was carried out by filtering catalyst (30% SiW11/MCM-41) from the reaction mixture after 4 h and allowed the filtrate to react up to 8 h. The reaction mixture of 4 h and filtrate was analysed by gas chromatography. Both the samples showed same conversion of 26%. It has been reported by Sheldon et al. that there are three categories for a catalyst to behave as a true heterogeneous catalyst in context of leaching of metal from the support (a) the metal leaches but is not active homogeneous catalyst, (b) metal leaches to form an active homogeneous catalyst and (c) the metal does not leach is a true heterogeneous catalyst. The results indicate that the present catalyst fall into category C.31 On the basis of these results, it can be concluded that there is no any leaching of the SiW11 from the support and the present catalysts are truly heterogeneous in nature. Similar results were obtained for 30% SiW12/MCM-41.3.6 Recycling study
The catalysts were recycled up to four times in order to test their activity in successive runs. The catalysts were separated from the reaction mixture by simple centrifugation, washed with 5 mL methanol and then with 5 mL distilled water, dried at 100 °C in an oven for 10 h and the recovered catalysts were charged for the further runs. The conversion of glycerol observed for four successive runs for 30% SiW11/MCM-41 are 62%, 60%, 58% and 57% and for 30% SiW12/MCM-41 are 75%, 72%, 71% and 70%. Thus, the catalysts can be reused up to four cycles with minimal loss in the conversion.Further the recycled catalysts were characterized by FT-IR analysis and BET surface area in order to see any structural change. The FT-IR spectrum of recycled 30% SiW12/MCM-41 showed the retention of typical bands for SiW12, at 979 cm−1 and 923 cm−1 corresponding to WOd and Si–Oa symmetric stretching, respectively. The FT-IR spectrum of the used catalyst 30% SiW11/MCM-41 (Fig. 10) shows retention of bands at 960 cm−1 (WOd), 900 cm−1 (Si–Oa) suggesting that the structure of SiW11 in regenerated catalyst is intact. The BET surface area values of the recycled catalysts (520 for 30% SiW11/MCM-41 and 332 for 30% SiW12/MCM-41) are comparable with the fresh ones (536 for 30% SiW11/MCM-41 and 349 for 30% SiW12/MCM-41).
 | ||
| Fig. 10 FT-IR spectra of fresh and recycled catalysts (a) 30% SiW12/MCM-41 and (b) 30% SiW11/MCM-41. | ||
3.7 Comparison with the reported catalysts
It can be seen from the Table 5 that all the reported catalysts operate at higher catalyst amount, high molar ratios and suffer from low conversion and selectivity.11,32,33 In contrast our catalysts prefer low Gly/urea ratio, requires very low catalyst amount and most importantly both conversion and selectivity are very high. The superiority of the present catalytic system lies in achieving very high activity and selectivity under moderately mild conditions.| Catalyst | Reaction conditionsa | Conv./Sel.b | Remark | Ref. |
|---|---|---|---|---|
| a Reaction conditions = amount of catalyst (mg): ratio of G/U: reaction temperature °C: reaction time (h).b Gly C selectivity. | ||||
| ZnSO4 | 250:1/1.5:150:4 |
83/58 | High catalyst amount | 11 |
| Au/TiO2 | 250:1/1.5:150:4 |
69/37 | Low selectivity | 11 |
| 2.5 wt% Au/MgO | 250:1/1.5:150:4 |
81/68 | High conversion and selectivity | 11 |
| PS-(Im)2ZnCl2 | 5 wt%:1/140:6 |
61/58 | Low selectivity | 32 |
| CeO2 | 60:1/1.5:140:1 |
24/96 | Low conversion | 33 |
| Nd2O3 | 60:1/1.5:140:1 |
30/92 | Low conversion | 33 |
| 30% SiW12/MCM-41 | 100:1/1:150:8 |
75/77 | High conversion and selectivity | This work |
| 30% SiW11/MCM-41 | 100:1/1:150:8 |
62/75 | High conversion and selectivity | This work |
3.8 Proposed mechanism for carboxylation reaction
Lingaiah et al. have reported a mechanism for glycerolysis of urea over Sm-TPA and we expect the same mechanism in the present case.18 The probable transesterification mechanism between triglyceride and methanol is shown in Scheme 2. Very high surface area and pore dimension of the present catalyst plays an important role in diffusion of large substrate molecules on to the strong acidic sites of the catalyst. The acidic sites of the catalysts facilitate the nucleophilic attack of glycerol oxygen to the carbonyl carbon of the urea. A tetrahedral intermediate is formed by nucleophilic attack of hydroxyl group of glycerol followed by subsequent release of ammonia molecules GlyC is formed. The leaving off of the product molecule is again a steric effect in the heterogeneous catalysis and the present catalyst contains large pore diameter that facilitates the escape of product molecule. | ||
| Scheme 2 Proposed mechanism for glycerolysis of urea. | ||
4. Conclusions
The reaction of glycerol with urea to synthesize GlyC is an attractive reaction that utilises two low-cost and easily available raw materials in a chemical cycle that, results in the fixation of carbon dioxide. Silicotungstates impregnated to MCM-41 have been found to be effective catalysts to promote the formation of GlyC. The catalyst, 30% SiW12/MCM-41 was found to be better amongst the two and activity was directly correlated with the higher acidity of the catalyst confirming a pseudo-liquid type (I) heterogeneous catalysis. The recycling study showed that both the catalysts could be reused with minimal loss in the activity. Further the FT-IR analysis confirms the retention of the structure of Keggin anion in the regenerated catalysts.Acknowledgements
Mr Nilesh Narkhede is thankful to Council of Scientific and Industrial Research (CSIR), New Delhi, for the award of Senior Research Fellowship.Notes and references
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434 CrossRef CAS PubMed.
- C. H. C. Zhou, J. N. Beltramini, Y. X. Fan and G. Q. M. Lu, Chem. Soc. Rev., 2008, 37, 527 RSC.
- N. Dimitratos, J. A. Lopez-Sanchez and G. J. Hutchings, Top. Catal., 2009, 52, 258 CrossRef CAS.
- A. C. Simao, B. Lynikaite-Pukleviciene, C. Rousseau, A. Tatibouet, S. Cassel, A. Sackus, A. P. Rauter and P. Rollin, Lett. Org. Chem., 2006, 3, 744 CrossRef CAS.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529 RSC.
- L. Ubaghs, N. Fricke, H. Keul and H. Hocker, Macromol. Rapid Commun., 2004, 25, 517 CrossRef CAS PubMed.
- V. Plasman and T. Caulier, Plast. Addit. Compd., 2005, 7, 30 CrossRef CAS.
- M. Aresta, A. Dibenedetto, F. Nocito and C. Ferragina, J. Catal., 2009, 268, 106 CrossRef CAS PubMed.
- M. A. Pacheco and C. L. Marshall, Energy Fuels, 1997, 11, 2 CrossRef CAS.
- A. Dibenedetto, A. Angelini, M. Aresta, J. Ethiraj, C. Fragale and F. Nocito, Tetrahedron, 2011, 67, 1308 CrossRef CAS PubMed.
- B. Hammond, J. A. Lopez-sanchez, A. R. Mohd Hasbi, N. Dimitratos, R. L. Jenkins, A. F. Carley, Q. He, C. J. Kiely, D. W. Knight and G. J. Hutchings, Dalton Trans., 2011, 40, 3927 RSC.
- J. R. Ochoa-Gomez, O. Gomez-Jimenez-Aberasturi, B. Maestro-Madurga, A. Pesquera-Rodriguez, C. Ramirez-Lopez, L. Lorenzo-Ibarreta, J. Torrecilla-Soria and M. C. illaran-Velasco, Appl. Catal., A, 2009, 366, 315 CrossRef CAS PubMed.
- M. J. Climent, A. Corma, P. De Frutos, S. Iborra, M. Noy, A. Velty and P. Concepcion, J. Catal., 2010, 269, 140 CrossRef CAS PubMed.
- J. B. Li and T. Wang, J. Chem. Thermodyn., 2011, 43, 731 CrossRef CAS PubMed.
- J. W. Yoo and Z. Mouloungui, Stud. Surf. Sci. Catal., 2003, 146, 757 CAS.
- F. R. Marcos, V. C. Casilda, M. A. Banares and J. F. Fernandez, J. Catal., 2010, 275, 288 CrossRef PubMed.
- J. H. Park, J. S. Choi, S. K. Woo, S. D. Lee, M. Cheong, H. S. Kim and H. Lee, Appl. Catal., A, 2012, 433–434, 35 CrossRef CAS PubMed.
- C. R. Kumar, K. Jagadeeswaraiah, P. S. S. Prasad and N. Lingaiah, ChemCatChem, 2012, 4, 1360 CrossRef CAS PubMed.
- K. Jagadeeswaraiah, C. R. Kumar, P. S. S. Prasad, S. Loridant and N. Lingaiah, Appl. Catal., A, 2014, 469, 165 CrossRef CAS PubMed.
- N. Narkhede and A. Patel, Ind. Eng. Chem. Res., 2013, 52, 13637 CrossRef CAS.
- M. Hinoa and K. Arata, Green Chem., 2001, 3, 170 RSC.
- B. Katryniok, S. Paul and F. Dumeignil, Green Chem., 2010, 12, 1910 RSC.
- N. Narkhede, S. Singh and A. Patel, Green Chem., 2015, 17, 89 RSC.
- E. Skliri, I. N. Lykakisb and G. S. Armatas, RSC Adv., 2014, 4, 8402 RSC.
- A. Patel and N. Narkhede, Catal. Sci. Technol., 2013, 3, 3317 CAS.
- N. Narkhede and A. Patel, RSC Adv., 2014, 4, 19294 RSC.
- P. Ferreira, I. M. Fonseca, A. M. Ramos, J. Vital and J. E. Castanheiro, Appl. Catal., B, 2010, 98, 94 CrossRef CAS PubMed.
- N. Narkhede and A. Patel, J. Porous Mater., 2014, 21, 579 CrossRef CAS.
- Y. Yang, Y. Guo, C. Hu and E. Wang, Appl. Catal., A, 2003, 252, 305 CrossRef CAS.
- G. Koyano, K. Ueno and M. Misono, Appl. Catal., A, 1999, 181, 267 CrossRef CAS.
- R. A. Sheldon, M. Walau, I. W. C. E. Arends and U. Schuchurdt, Acc. Chem. Res., 1998, 31, 485 CrossRef CAS.
- D.-W. Kim, K.-A. Park, M.-J. Kim, D.-H. Kang, J.-G. Yang and D.-W. Park, Appl. Catal., A, 2014, 473, 31 CrossRef CAS PubMed.
- L. Wang, Y. Ma, Y. Wang, S. Liu and Y. Deng, Catal. Commun., 2011, 12, 1458 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |