
Recent advances in graphene/polyamide 6 composites: a review
Abstract
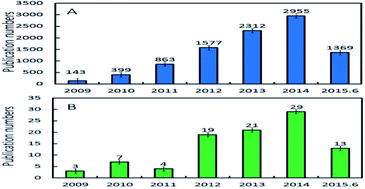
This paper reviews recent advances in polyamide 6 (PA6) nanocomposites with graphene-based fillers including current works using graphite nanoplatelet fillers. Almost all of the latest important publications relating to the preparation, morphology and properties (such as thermal stability, thermal conductivity, electrical conductivity, mechanical properties, and flame retardant and gas barrier properties) of graphene/PA6 nanocomposites are summarized. An outlook of the current challenges in this field is provided for a potential guide to progress in the development of graphene/polymer nanocomposites too.
 Please wait while we load your content...
Please wait while we load your content...

