
Role of lithium oxide as a sintering aid for a CGO electrolyte fabricated via a phase inversion technique
Siti Munira Jamil,
Mohd Hafiz Dzarfan Othman*,
Mukhlis A. Rahman,
Juhana Jaafar,
Ahmad Fauzi Ismail and
Mohamad Azuwa Mohamed
Advanced Membrane Technology Research Centre (AMTEC), Universiti Teknologi Malaysia, 81310 UTM Johor Bahru, Johor Bahru, Malaysia. E-mail: hafiz@petroleum.utm.my; Fax: +607-5535925; Tel: +607-5536373
First published on 26th June 2015
Abstract
The incorporation of lithium oxide (Li2O) as a sintering additive has specific advantages for electrolyte membrane fabrication. However, the viability of the sintering additive to be implemented in a phase inversion technique is still ambiguous. In this first attempt, lithium was doped into a gadolinium-doped ceria (CGO) crystal structure using the metal nitrate doping method and calcined at four different temperatures, i.e. 140, 300, 500 and 700 °C. The prepared Li-doped CGO (Li–CGO) powders were analyzed by thermal gravimetric analysis (TGA), differential scanning calorimetry (DSC), X-ray diffraction (XRD), N2 adsorption/desorption, and Fourier-transform infrared (FTIR). Primary results demonstrate that the calcination temperature of the Li–CGO influences the condition of the electrolyte suspension. Li–CGO calcined at 700 °C (D-700), as compared with other Li–CGO, possessed a strong interaction between the Li and CGO. The D-700 was then incorporated into the electrolyte flat sheet membrane which was prepared by a phase inversion technique. The membrane was then sintered at different sintering temperatures from 1350 °C to 1450 °C. In comparison with the unmodified CGO, the morphological results suggest that the Li2O can remarkably promote the densification of CGO at a lower sintering temperature (1400 °C). These findings help to promote the use of sintering additives in a ceria-based electrolyte suspension specifically for the phase inversion technique.
1. Introduction
A solid oxide fuel cell (SOFC) is an energy converter, from chemical to electrical through fuel oxidizing. As a green alternative to depreciating fossil fuel resources, it is highly efficient, low in emissions, and flexible in fuel selection. To date, intermediate temperature SOFCs (IT-SOFC) which operate within 400–700 °C are preferable to high temperature SOFCs (HT-SOFC) which operate within 700–1000 °C, due to their long-term durability and cost effectiveness. Electrolytes for SOFCs are mainly based on yttrium-doped zirconia (YSZ) and cerium–gadolinium oxide (CGO). It has been widely reported that CGO possesses four to five times higher conductivity at intermediate operating temperatures,1–7 which qualifies its compatibility for IT-SOFC. However, sintering temperatures as high as 1550 °C are typically needed to densify the CGO electrolyte8 which eventually increases the cost and complicates the cell fabrication process. Therefore, densification of CGO electrolyte at lower temperature has become the main focus of SOFC research recently.Generally, the fabrication of ceramic membrane precursor starts with the formation of suspension, followed by shaping the suspension into electrolyte precursor in a form of flat sheet or tubular shape. The last step is the consolidation of the electrolyte precursor by sintering.9 Currently, phase inversion method has been the most effective in the fabrication of SOFC.10 It allows flexible and micro-scale control on the internal structure of the membrane precursor. Since last decade, many researchers have attempted to reduce the sintering temperature of densified CGO. The first method is by introducing metal oxides as the sintering aids to get fully dense CGO at 900–1200 °C (ref. 11–13) and another is by using particle of nano size as the starting powder.14,15
To date, adding a small amount of sintering additive has been found effective to reduce the densification temperature. For example, Nicholas and Jonghe doped 3 mol% of Li2O into CGO layer and was able to achieve up to 99% of relative density of Ce0.9Gd0.1O1.95 after 800 °C of sintering.16,17 Another study reported similar results but with lower amount of Li2O additive which was 2.5 mol%.18 Nevertheless, the previous works only reported on CGO button cells prepared by pressing method. To the best of author's knowledge, study on the effect of Li2O as sintering additive on electrolytes prepared by phase inversion technique, has yet been reported. The feasibility of the sintering additive on this technique is still unclear.
According to literatures, most CGO powders were mixed with Li salt or oxide. After that, the mixed powders were calcined and pressed to form pellets. The pellets were sintered for further solidification. They were then characterized in term of microstructure and electrical properties. The calcination temperature could vary from 130 to 600 °C.13,16,18,19 In our previous study, the NiO–CGO/Li–CGO dual-layer hollow fiber precursors was co-sintered at 1150 °C,20 in which the preparation of Li–CGO powders was adopted from Nicholas16 prior to the deposition of electrolyte layer by brush painting.
However, the preparation of the electrolyte suspension for phase inversion could be more complicated when it involves the incorporation of sintering additives. Such suspension which contains Li–CGO requires certain rheology to ensure the attachment between the Li and CGO sites consistently strong. This is to prevent the Li from leaching out of the CGO crystal structure otherwise, it will disrupt the suspension stability and solidification during phase inversion. Thus, it is crucial to understand the effect of calcination temperature on the use of sintering additive in phase inversion technique.
The objective of this study is to determine the optimum calcination temperature of Li–CGO powder to satisfy the stability of the electrolyte suspension during phase inversion. The Li–CGO powder was calcined at 140, 300, 500, and 700 °C. The powder was characterized by TGA, DSC), XRD, N2 adsorption/desorption, and FTIR. The feasibility of using Li2O as sintering additive in CGO for phase inversion was evaluated based on the Li–CGO morphological structures by scanning electron microscopy (SEM) images after the precursor was sintered at 1350 °C to 1450 °C.
2. Experimental
2.1 Materials
Commercially available nano sizes cerium–gadolinium oxide powder (Ce0.9Gd0.1O1.95 or CGO, surface area 192 m2 g−1, particle size −60 mesh d50) was purchased from NexTech Materials Ltd (Ohio) and used as received. Lithium nitrate (LiNO3) (AR Grade, QRec) was used as the sintering additive. Polyethersulfone (PESf) (Radel A-300, Ameco Performance) and polyethylene glycol 30-dipolyhydroxystearate (PEG) (Arlacel P135, Uniqema) was used as the polymer binder and dispersant, respectively. N-methyl-2-pyrrolidone (NMP) (HPLC grade, Rathbone) was used as the solvent for electrolyte suspension and tap water as coagulant.2.2 Doping of CGO with Li (Li–CGO)
The electrolyte material was doped with the sintering additives, LiNO3 via metal nitrate doping method. The loading of the additive was 2 mol% which was within the common range of 0.5–3 mol%.4,16 The Li–CGO was prepared by uniformly mixing 2 mol% of LiNO3 solution with the CGO powder. Then, the mixture was heated at 80 °C until dry. After that, the dried powder was ground prior to calcination at 140 (D-140), 300 (D-300), 500 (D-500), and 700 °C (D-700) for 2 hours with 5 °C min−1 of heating rate to oxidize the nitrates and turn into surface dopants.2.3 Electrolyte suspension and flat sheet preparation
The suspension prepared is typically a viscous mixture composed of ceramic particles, solvent, binder and additives, which each contributes on the physical and chemical properties of the suspension. The particle size, distribution and shape are factors that affect the dispersion of ceramic particles in the suspension and its corresponding precursor properties. The amount of binder should be kept at minimum to prevent any noticeable effects on the phase inversion. Additionally, they should be soluble in the precursor solvent and cleanly burn-off during sintering. High exchange rate between the solvent and the non-solvent (coagulants) is usually preferable. In fact, the rates of solvent outflow and coagulant inflow affect the final morphology of the membrane formed.9Li9 has highlighted four points to consider before preparing ceramic suspension: (1) the amount of dispersant to stabilize the suspension; (2) the minimum amount of solvent to homogenize the suspension; (3) the minimum ratio between organic components and ceramic powders (4) the ratio of plasticizer to binder to make the membrane precursor flexible, resistive and shapeable.
The Li–CGO powder prepared earlier was first mixed with solvent and dispersant. The mixture was milled for 48 h using NQM-2 planetary ball miller, Magna value and polymer binder was slowly added into the mixtures during the milling. The milling was continued for another 48 h to homogenize the suspension. Prior to casting, the electrolyte suspension was stirred at room temperature to degas for 1 h. The suspension was then casted on a glass plate using glass rod to form a thin film. The thin film was immersed overnight to complete the phase inversion process and dried under ambient condition for 24 h to form a membrane. Similar membrane was produced but using undoped CGO for comparison study.
Finally, the membrane precursors were sintered inside a tube furnace (XY-1700, Magna value). The temperature was increased from room temperature to 400 °C at 5 °C min−1 for 1 h, then to 800 °C at 5 °C min−1 for 1 h and finally to target temperature (i.e. 1350, 1400 and 1450 °C) at 10 °C min−1 for 12 h. The temperature was then reduced to room temperature at 5 °C min−1.
2.4 Characterizations
Thermal decomposition of LiNO3 and Li–CGO powders was analyzed by TGA (Netsch STA 449C) using nitrogen as carrier gas. The sample weight, heating rate and final temperature range were 5 mg, 10 °C min−1, and 1000–1200 °C, respectively. In addition, the thermal transformations (melting) of LiNO3 and Li–CGO powders were analyzed by DSC (Netsch STA 449C). The carrier gas was nitrogen and the final temperature was 600 °C.High resolution X-ray diffractometer (Bruker D8 Advance) analyzer was used to identify the crystal phases of the Li–CGO powders. The XRD analysis was applied using a 40 kV 40 mA X-ray tube, employing a CuKα radiation at a wavelength of 2.146 nm, 0.5° divergence and anti-scatter slits, and a 0.1 mm receiving slit. The sintered samples were analyzed in the range of 10 ≤ 2θ (°) ≤ 80 at 1° of scan rate. The average size of the Li–CGO crystallite was estimated using Scherrer equation.21
 | (1) |
The specific surface area of particles was determined using the N2 adsorption/desorption (Pulse Chemi Sorb 2705) and analyzed using Brunauer–Emmett–Teller (BET). The surface area is determined by the amount of adsorbed gas at a given pressure. Fourier-transform infrared (FTIR) spectra of the Li–CGO were recorded on IRTracer-100 Shimadzu spectrophotometer. The spectra were recorded at resolution between 350 and 4000 cm−1.
Finally, the morphology of the flat sheet was examined using scanning electron microscopy (SEM) (TM3000, Hitachi) at an accelerating voltage of 15 kV. The flat sheets were snapped in order to expose their cross-sectional fractures. Then, they were placed on a metal holder and gold-sputtered under vacuum. High resolution images of the cross-section and surface of the flat sheets was taken at different magnifications.
3. Results and discussion
3.1 Thermal decompositions of LiNO3 and Li–CGO powders
Transformations mechanism during calcination of pure LiNO3 and Li–CGO powders were studied by TGA and DSC analyses. Fig. 1 shows the results of TGA and DSC of LiNO3. As can be seen, the LiNO3 salts started to melt at 251.77 °C (curve (b)) and decompose at 320.83 °C (curve (a)). Similar pattern was observed by Ruiz et al.22 They reported that the decomposition of LiNO3 could turn into either Li2O at 650 °C or lithium peroxide (Li2O2) above 650 °C, and both were accompanied by the release of nitrogen dioxide. | ||
| Fig. 1 TG and DSC curves of LiNO3. | ||
Moreover, the TG curve of LiNO3 depicted that the weight decreased with temperature and the total weight loss after 1000 °C was almost 99.1%. These results suggest that the Li2O or Li2O2 will fully evaporate after 1000 °C. As reported by Zhu et al.,19 the high vapor pressure of Li2O (e.g. 1.09 × 10−8 bar) confirms that the Li2O can be developed as a non-residual sintering additive.
Fig. 2 shows the DSC diagrams of Li–CGO powders calcined at different temperatures. The melting signal was observed at 250 °C for the D-140, D-300, and D-500 which is close to LiNO3 as previously shown in Fig. 1. All samples were preheated at four different calcination temperatures; 140, 300, 500 and 700 °C. When the samples showed the same properties as LiNO3 which proven by DSC analysis, these results indicate that LiNO3 does not undergo any solid-state reactions with the CGO.22 In contrast, the D-700 diagram showed lower melting point which was 182 °C instead of around 250 °C like the others. These results suggest that the LiNO3 either is highly dispersed on the surface of CGO or strongly interacts with it.
 | ||
| Fig. 2 DSC of Li–CGO at different calcination temperature. | ||
Fig. 3 shows the TGA diagrams of LiNO3, CGO and Li–CGO powders at different calcination temperatures. In overall, the D-140 and D-300 lost their weights by two stages. However, three stages were observed for the D-500 and D-700. The first stage at approximately at 100 °C are corresponds to the evaporation of the physically adsorbed water. The second stage at 500–550 °C may be attributed to oxidation of LiNO3 and released of NO2 and O2. However, the oxidation occurred earlier for pure LiNO3 from 320 °C to 500 °C. The third stage at 880 °C may be attributed to evaporation of Li2O that was probably formed in the powders. Since LiNO3 start to decompose at 320 °C and boil at 600 °C,23 the Li2O will start to form liquid phases with Gd2O3 and CeO2 (Li–Gd–Ce–O)19 at this stage. As the temperature increased, the molten phase of Li2O evaporated.
 | ||
| Fig. 3 TGA diagrams of LiNO3, CGO and Li–CGO at different calcination temperature. | ||
Moreover, while LiNO3 almost completely decomposed at 1000 °C, none of the Li–CGO powders displayed similar behavior. These results suggest that the doped LiNO3 oxidizes and turns into Li2O which later interacts with the CGO sites. A series of Li–Gd–Ce–O phases with different compositions and lattice parameters may have been formed during heating up and it should be noted that the molten phase of Li2O was not completely evaporated at 1200 °C.
3.2 Effect of calcination temperature on the crystallinity of Li–CGO powders
Fig. 4 shows the XRD patterns of the LiNO3, CGO, and Li–CGO powders at different calcination temperatures. The peaks of the Li–CGO diffractograms became sharper with increased calcination temperatures. This is due to higher crystallinity and larger crystallite size of Li–CGO caused by higher heat energy supplied into the system resulting particle aggregations. In addition, these results also indicate that adding LiNO3 into CGO does not produce any new phases or induce any detectable structural changes in the CGO. | ||
| Fig. 4 X-ray diffractograms of Li–CGO powders compared with LiNO3 and CGO. | ||
3.3 Effect of calcination temperature on the surface area of Li–CGO
The BET method was used to determine the specific surface area of Li–CGO calcined at different temperatures. The data of pure CGOs with micron and nano size were presented for controls. As shown in Table 1, the surface area of Li–CGO powders decreased with increased calcination temperatures. The D-140 showed the highest surface area, 54.18 m2 g−1. As the temperature of calcinations was raised to 700 °C (D-700), the surface area decreased to 2.9 m2 g−1. This can be attributed to the thermally promoted crystallite growth.24| Powder | Surface area (m2 g−1) | Crystallite size by Scherrer equation (nm) |
|---|---|---|
| Pure CGO micron size | 7 | 427 |
| Pure CGO nano size | 212 | 76 |
| D-140 | 54 | 73 |
| D-300 | 53 | 74 |
| D-500 | 19 | 271 |
| D-700 | 3 | 691 |
Similar trend was also reflected by their crystallite sizes which increased with calcination temperatures. The size of the Li–CGO crystallites increased from 73.1 nm to 691.09 nm when the calcination temperature was raised from 140 to 700 °C. At 140 °C, the crystallite size was 73.10 nm and slightly increased to 74.15 nm at 300 °C. Apparently in this case, the surface areas can be correlated to the crystallite sizes, i.e. smaller crystallite size indicates larger specific surface area.
3.4 Effect of calcination temperature on the chemical interaction in Li–CGO powders
The effect of calcination temperature on the chemical interaction was investigated by observing the functional groups of the Li–GCO powder using FTIR analysis. The FTIR spectra of raw CGO, LiNO3 and Li–CGO at different calcination temperatures are shown in Fig. 5. The frequency and assignment of each vibrational mode observed are listed in Table 2. As shown in the Fig. 5, all samples showed absorption band at 3000–3600 cm−1 and 1640 cm−1 representing O–H stretching of hydroxyl group and the vibration of adsorbed water on the surface of the samples.25,26 | ||
| Fig. 5 FT-IR spectrum of LiNO3, CGO and Li–CGO powders at different calcination temperature. | ||
| Peak assignment | Peak frequency (cm−1) | |||||
|---|---|---|---|---|---|---|
| CGO | LiNO3 | D-140 | D-300 | D-500 | D-700 | |
| O–H stretching of hydroxyl groups | 3852 | — | 3853 | — | — | — |
| 3739 | — | — | — | — | — | |
| 3450 | 3450 | 3450 | 3417 | 3417 | — | |
| LiO–H stretching | — | — | — | — | — | 3564 |
| C–C triple bonds | 2359 | — | 2359 | 2358 | 2358 | 2360 |
C–O and C–C double bonds conjugation (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O−)3Gd and (OC–O−)3Ce stretching vibration C–O−)3Gd and (OC–O−)3Ce stretching vibration |
1630 | 1633 | 1630 | 1637 | 1637 | 1570 |
| O–H stretching antisymmetric N–O vibration mode of free nitrate ions | 1385 | 1384 | 1384 | 1325 | 1328 | 1436 |
| 857 | 823 | 857 | 823 | 862 | 860 | |
| C–O single bond | 1100 | — | 1100 | 1100 | 1100 | 993 |
| Vibration of C–H |
1069 | — | 1069 | — | 1087 | — |
| Vibration of C–H |
1052 | — | 1050 | — | 1049 | — |
Moreover, the presence of absorption band at 1625–1650 cm−1 relates to (OC–O−)3Gd and (OC–O−)3Ce stretching vibration.24 Besides, all modified samples show the presence of anti-symmetric N–O vibration mode of free nitrate ions at 1347–1384 cm−1 and 828–850 cm−1.22,27,28 The intensity of these absorption bands decreased as the calcination temperature was increased up to 700 °C. The disappearance of 1640 cm−1 absorption band for sample D-700 indicates the removal of water from the surface of the samples as the water vaporizes at high temperature. The sharp absorption band at 3564 cm−1 observed in D-700 is attributed to hydroxyl stretching vibration of Li–O–H.29
Additionally, small shoulder absorption was also observed such as the band at 734–739 cm−1 which represents the vibrational stretching of lithium compound (Li–O).30 Several other notable absorption peaks of low intensity were also observed including peaks at 1050 cm−1, 1110 cm−1, 1630 cm−1, and 2359 cm−1 corresponding to vibrational C–H, C–O single bond, C–O and C–C double bonds conjugation, and C–C triple bonds, respectively.26
Therefore, these FTIR analyses confirm that the nitrates is oxidized as surface dopants. It is clear based on the appearance of hydroxyl stretching vibration of Li–O–H and vibrational stretching of new lithium compound (Li–O) in the D-700. This argument is also supported by the decrease of anti-symmetric N–O vibration mode of free nitrate ions absorption band. In addition, these results are in accordance with the TGA result of D-700. Consequently, this result confirms the formation of new lithium compound of Li–O in the D-700 powders.
3.5 Effect of sintering additive on electrolyte suspension
As mentioned in Section 2.2, the properties of ceramic powder could affect its dispersion in the suspension and properties of the electrolyte precursor. From the TGA and DSC results, it is assured that the addition of LiNO3 as sintering additive into CGO has successfully produced Li–CGO powders at 700 °C of calcination temperature. Their characteristics are believed to be stable as it has almost the same TGA curve as the undoped CGO. Furthermore, the crystallite size and surface area of D-700 powders represent the contiguous data as the micron size CGO. Even though higher surface area is good to improve the densification of CGO, moderate surface area like micron size CGO is preferable for electrolyte preparation via phase inversion technique. This is because, in dope suspension, polymer binder is needed to bind the ceramic particle during solvent–non-solvent exchange process. Thus, higher surface area requires higher amount of polymer binder to keep the ceramic particles intact. Besides, powder with high surface area tends to induce agglomeration. Additionally, the FTIR results confirm the existence of the desired new compound Li–O in D-700 powders. Therefore, the D-700 powder was selected to prepare the electrolyte suspension.Despite that, the electrolyte suspension was also prepared using D-140 and D-500 powders in order to justify their incompatibility with the phase inversion technique. The compositions of the suspension are listed in Table 3 as well as the observations after immersing the flat sheet precursor in water. If the ceramic loading was the same as neat suspension (D-0) which was 58%, it resulted in undissolved polymer binder, which was polyethersulfone (PES). There is a slight possibility that the suspension is affected by the characteristics of the salt–solvent medium itself whereby Li interaction with the aprotic solvents (e.g. NMP) is stronger than its interaction with PES polymer chains. The same phenomena was also observed by Iqbal et al.31 The reduction in solvent power contributes to the insolubility of PES. Therefore, the ceramic loading for suspension with Li content (D-140, D-500 and D-700) must be maintained at 50% to ensure high homogeneity and the inversion capability of the suspension remains consistent.
| Suspension | CGO | Li–CGO | PESf | Arlacel P135 | NMP | Observation |
|---|---|---|---|---|---|---|
| Undoped CGO (D-0) | 58.00 | — | 5.80 | 0.12 | 36.08 | Good rheology |
| Doped CGO (D-140) | — | 58.00 | 5.80 | 0.12 | 36.08 | Undisssolved PESf |
| Doped CGO (D-140) | — | 50.00 | 5.00 | 0.12 | 44.88 | Homogeneous but white spot appear on the surface of water when immersed |
| Doped CGO (D-500) | — | 58.00 | 5.80 | 0.12 | 36.08 | Undisssolved PESf |
| Doped CGO (D-500) | — | 50.00 | 5.00 | 0.12 | 44.88 | Homogeneous but white shadow emerge on the glass plate when immersed |
| Doped CGO (D-700) | — | 58.00 | 5.80 | 0.12 | 36.08 | Undisssolved PESf |
| Doped CGO (D-700) | — | 50.00 | 5.00 | 0.12 | 44.88 | Homogeneous and solidified during phase inversion |
However, when the D-140 and D-500 flat sheets were immersed in water, some white particles leached out from the flat sheets as shown in Fig. 6(a) and (b). This phenomenon may be attributed to the low degree of crystallinity of Li–CGO powders with the presence of amorphous phase, making the structure unstable and leached out when immersed in water. Moreover, it is important to note that the stability of the crystalline structure depends on the internal bonds between the molecules in the crystal lattice.32 As a result, at low calcination temperature, the Li ions might not strongly attach to the CGO sites.
 | ||
| Fig. 6 (a) D-140 and (b) D-500 flat sheets while immersing in water. | ||
On contrary, a well-shaped flat sheet was obtained for D-700. As proven by the TGA, DSC, XRD, N2 adsorption/desorption and FTIR analyses, D-700 possesses distinctive characters as compared with the other doped CGO powders, which indicates strong interaction was established between CGO–Li when the powder was calcined at 700 °C. This observation demonstrates that the D-140 and D-500 powders are not suitable for electrolyte preparation via phase inversion technique and the D-700 powder meets better requirement for electrolyte suspension preparation.
3.6 Effect of sintering additive on microstructure of CGO electrolyte
Fig. 7 shows SEM image of the flat sheet developed in this study that was sintered at 1350 °C. The cross-section of the flat sheet exhibits the symmetric structure as the sponge-like structure occupying the flat sheet. The formation of finger-like structure was fully suppressed by drying the flat sheet in ambient before immersing in water which then making the local viscosity of the flat sheet exceeds a critical level.33 Thus, the viscous fingering phenomenon was hindered and no finger-like structure was observed. The thickness of the flat sheet was in the range from 260 to 130 μm as the thickness decreases with the increased of sintering temperature. | ||
| Fig. 7 SEM images of the full cross-section of the flat sheet. | ||
To further analyze the effect of sintering additive on the microstructure of CGO electrolyte, analysis on the cross sections and surfaces images taken by the SEM was performed. The precursor from D-700 was sintered and compared with the undoped precursors as shown in Fig. 8 for cross section images and Fig. 9 for surface images. For cross section images, the zoomed area was focused at the centre region of the flat sheet. The effect of sintering additive in CGO can be clearly seen in terms of sintering temperature and the role of sintering additive itself. The result indicates that the sintering process is enhanced at 1350 to 1450 °C, resulting in early densification of CGO. As can be seen, the Li–CGO sintered at 1350 °C is denser than the unmodified CGO. Eventually, with only 2 mol% of LiNO3 doping in CGO, the densification process was completed at 1400 °C. In comparison, the undoped CGO might require up to 1550 °C to achieve similar level of solid state sintering.33 The result indicates that Li2O as sintering additive can remarkably promote the densification of CGO at sintering temperature as low as 1400 °C.
 | ||
| Fig. 8 SEM cross sections images of undoped CGO and Li–CGO at different sintering temperatures. | ||
 | ||
| Fig. 9 SEM surface images of undoped CGO and Li–CGO at different sintering temperatures. | ||
Furthermore, in order to maintain the conductivity in SOFC application, non-residual sintering aids are most preferred.34 Li2O was reported to be a highly efficient sintering aid for CGO as up to now, it has the highest vapor pressure among all the studied metal oxides.19 This will ensure the complete evaporation of Li2O during sintering. In fact, various mechanisms have been proposed to explain the sintering mechanism of Li doped CGO precursor, such as beneficial liquid formation, dopant substitution, and dopant segregations on the grain boundaries. Kleinlogel and Gauckler35,36 attributed the enhanced conductivity to the electronic conduction due to segregation or dissolution of heavy metal oxides on the grain boundaries. In contrast, Esposito et al.23 believed that the added salt LiNO3 melts and evaporates in large part during sintering, and thus enhances the ionic conductivity especially at the grain boundaries. A number of researchers clarified that the Li2O facilitates the sintering by way of a liquid phase sintering mechanism.13,18,19,34,37 In this study, the role of Li2O that facilitates the early densification of CGO at 1400 °C may be attributed to liquid phase sintering as well.
To further elucidate, during the formation of molten phase of Li2O at temperature 880 °C as defined by TGA analysis, the Li ions probably enter the lattice of CGO to form Li–Gd–Ce–O phases. As described by Zhu et al.,19 Li diffused into the CGO lattice and vaporized from the lattice as the sintering temperature increased. It is probable that Li doped in both Gd and Ce sited as shown in the following reactions:
 | (2) |
 | (3) |
Consequently, the Li–CGO liquid possibly forms on the surface of the CGO grains during sintering which leads to a disorder and increased activity on the surface of CGO grains as depicted in Fig. 10. Then, Li2O adsorbs onto the grain surface of CGO to form a thin layer promoting atomic diffusion rate between grain boundaries for rearrangement. This mechanism lowers the net energy requirement for the structure densification which eventually reduces the sintering temperature. In fact, these mechanism were correspond to the structural evolution of metal oxide in CGO that was enlightened by Zhang et al.38 They indicates that Ni can be incorporated into CGO lattice in three ways; (1) substitution of metal ions to the Ce4+/Gd3+ fluorite structure, (2) formation of metals–CGO solid solution at the interstitial sites due to large difference in ionic radius between Ni2+ (0.69 Å) and Ce4+/Gd3+ (0.97/1.053 Å), (3) metals dispersed on the surface of CGO particles. As both Ni and Li were classified as metals, almost similar mechanism could be predicted for the Li–Gd–Ce–O system in this work. However, with the small difference in ionic radius between Li+ (0.92 Å) and Ce4+/Gd3+ (0.97/1.053 Å), it is expected that the reaction took place beneficially at grain boundary regions and migrated further due to the vaporization of Li2O on the surface of the sintered flat sheet as Li2O is much more evaporable compared to NiO.19
 | ||
| Fig. 10 Liquid phase sintering. | ||
Ultimately, at 1400 °C sintering temperature, Li2O was expecting completely vaporized. As correlated by XRD analysis in Fig. 11, the peaks of D-700 sintered at 1400 °C demonstrating the most similar lattice parameter for all samples as it was found to be almost the same as undoped CGO and Li doped CGO. This is in accordance with a simulation study that has been carried out by Burbano et al.39 to examined the effect of cations co-doping in ceria oxide (CeO2). The outcomes demonstrated that co-doping would not change the host lattice. In other words, it could duplicate the lattice constant of ceria doped with a cation which has an ionic radius equivalent to the effective radius from two co-dopants.
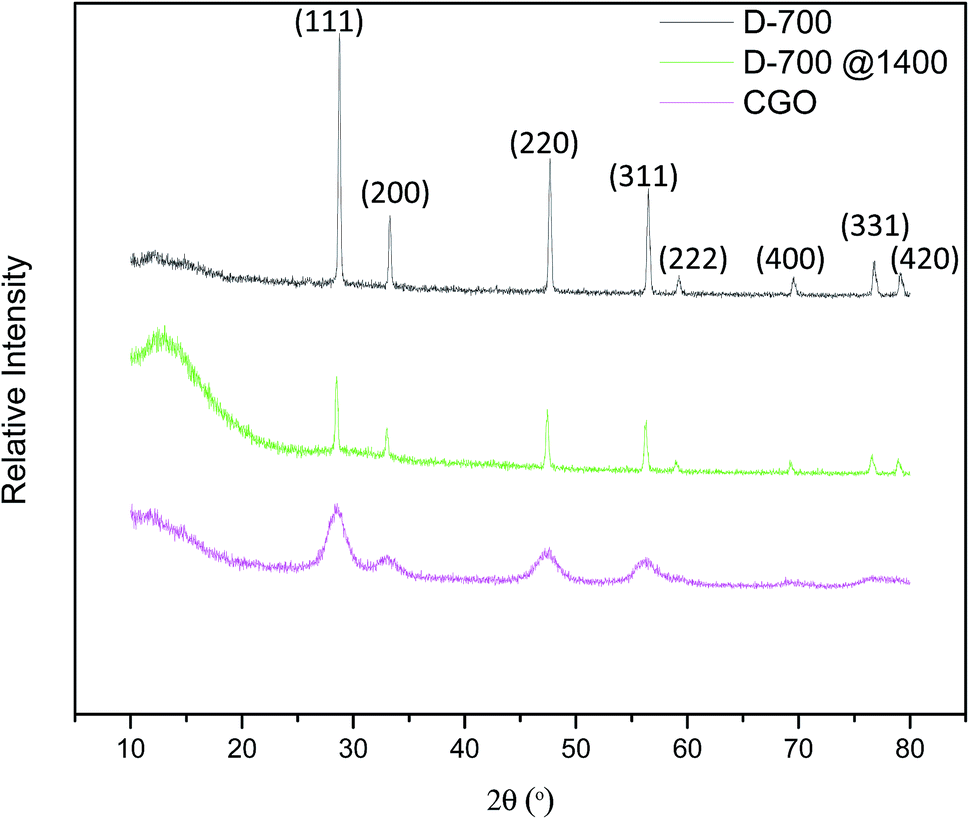 | ||
| Fig. 11 X-ray diffractograms of D-700 flat sheet sintered at 1400 °C compared with D-700 and CGO. | ||
4. Conclusions
The sintering temperature to synthesize dense CGO electrolyte layer has been successfully lowered by incorporating Li2O as sintering additive. The sintering temperature was lowered to 1400 °C by doping 2 mol% of LiNO3 into CGO via metal nitrate doping method. In addition, the calcination temperature of Li–CGO during powder preparation plays an important role as well. That is, the Li–CGO powders must be calcined at 700 °C to produce the desired rheology of electrolyte suspension for it to be compatible with the phase inversion technique. It can be concluded that Li2O doped Ce0.9Gd0.1Oδ electrolyte is a promising candidates for SOFCs at intermediate temperature. With the utilization of low sintered electrolyte, low processing cost and time as well as improved quality of SOFCs component can be produced. This study has proven the possibility to accelerate the densification by incorporating sintering additives as well as presented an alternative solution to reduce the cost in SOFC fabrication process.Acknowledgements
The authors gratefully acknowledge financial support from Ministry of Education Malaysia under Exploratory Research Grant Scheme (Project number: R.J130000.7842.4L104) and Fundamental Research Grant Scheme (Project number: R.J130000.7809.4F282), and Universiti Teknologi Malaysia under Research University Grant Tier 1 (Project number: Q.J130000.2509.05H53). The authors would also like to thank Research Management Centre, Universiti Teknologi Malaysia for the technical support.Notes and references
- A. Kaiser, A. S. Prasad, S. P. Foghmoes, S. Ramousse, N. Bonanos and V. Esposito, J. Eur. Ceram. Soc., 2013, 33, 549–556 CrossRef CAS PubMed.
- M. Chen, B. Hallstedt, A. N. Grundy and L. J. Gauckler, J. Am. Ceram. Soc., 2003, 86, 1567–1570 CrossRef CAS PubMed.
- D. P. Fagg, V. V. Kharton and J. R. Frade, J. Electroceram., 2002, 9, 199–207 CrossRef CAS.
- D. P. Fagg, J. C. C. Abrantes, D. Pérez-Coll, P. Núñez, V. V. Kharton and J. R. Frade, Electrochim. Acta, 2003, 48, 1023–1029 CrossRef CAS.
- D. P. Fagg, V. V. Kharton and J. R. Frade, J. Solid State Electrochem., 2004, 8, 618–625 CrossRef CAS PubMed.
- V. Gil, J. Tartaj, C. Moure and P. Durán, J. Eur. Ceram. Soc., 2006, 26, 3161–3171 CrossRef CAS PubMed.
- L. Zhang, R. Lan, P. I. Cowin and S. Tao, Solid State Ionics, 2011, 203, 47–51 CrossRef CAS PubMed.
- K. Maca, V. Pouchly and P. Zalud, J. Eur. Ceram. Soc., 2010, 30, 583–589 CrossRef CAS PubMed.
- K. Li, Ceramic Membranes for Separation and Reaction, John Wiley & Sons Ltd, England, 2007 Search PubMed.
- S. M. Jamil, M. H. D. Othman, M. A. Rahman, J. Jaafar, A. F. Ismail and K. Li, J. Eur. Ceram. Soc., 2014 Search PubMed.
- D. Perezcoll, P. Nunez, J. Abrantes, D. Fagg, V. Kharton and J. Frade, Solid State Ionics, 2005, 176, 2799–2805 CrossRef CAS PubMed.
- M.-F. Han, S. Zhou, Z. Liu, Z. Lei and Z.-C. Kang, Solid State Ionics, 2011, 192, 181–184 CrossRef CAS PubMed.
- S. Le, S. Zhu, X. Zhu and K. Sun, J. Power Sources, 2013, 222, 367–372 CrossRef CAS PubMed.
- J. A. Glasscock, V. Esposito, S. P. V. Foghmoes, T. Stegk, D. Matuschek, M. W. H. Ley and S. Ramousse, J. Eur. Ceram. Soc., 2013, 33, 1289–1296 CrossRef CAS PubMed.
- V. Gil, J. Gurauskis, R. Campana, R. I. Merino, a. Larrea and V. M. Orera, J. Power Sources, 2011, 196, 1184–1190 CrossRef CAS PubMed.
- J. D. Nicholas and L. D. Jonghe, Solid State Ionics, 2007, 178, 1187–1194 CrossRef CAS PubMed.
- Q. Yang and J. D. Nicholas, Effect of Sintering Aids on the Stress Evolution of Constrained Sintered Gadolinium-Doped Ceria Films, 2012 Search PubMed.
- M. Han, Z. Liu, S. Zhou and L. Yu, J. Mater. Sci. Technol., 2011, 27, 460–464 CAS.
- T. Zhu, Y. Lin, Z. Yang, D. Su, S. Ma, M. Han and F. Chen, J. Power Sources, 2014, 261, 255–263 CrossRef CAS PubMed.
- K. Manakor, S. Munira, M. Hafiz, D. Othman, M. A. Rahman, J. Jaafar and A. Fauzi, J. Teknol., 2014, 2, 33–39 Search PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- M. L. Ruiz, I. D. Lick, M. I. Ponzi, E. R. Castellón, A. Jiménez-López and E. N. Ponzi, Thermochim. Acta, 2010, 499, 21–26 CrossRef CAS PubMed.
- V. Esposito, M. Zunic and E. Traversa, Solid State Ionics, 2009, 180, 1069–1075 CrossRef CAS PubMed.
- Y. Chen, C. Lee, M. Yeng and H. Chiu, J. Cryst. Growth, 2003, 247, 363–370 CrossRef CAS.
- Z. Wang, G. M. Kale and M. Ghadiri, Chem. Eng. J., 2012, 198–199, 149–153 CrossRef CAS PubMed.
- J. M. G. Martínez, R. A. M. Meneses and C. R. M. da Silva, Mater. Sci. Forum, 2014, 798–799, 182–188 CrossRef.
- M. Sulaiman, N. A. Dzulkarnain, A. A. Rahman and N. S. Mohamed, Solid State Sci., 2012, 14, 127–132 CrossRef CAS PubMed.
- M. L. Ruiz, I. D. Lick, M. I. Ponzi and E. N. Ponzi, Catal. Commun., 2013, 34, 45–51 CrossRef CAS PubMed.
- A. S. S. Cavanagh, Y. Lee, B. Yoon and S. M. M. George, ECS Trans., 2010, 33, 223–229 CAS.
- I. M. Z. Rappoport, The chemistry of organolithium compounds Part 1, John Wiley & Sons, Ltd, West Sussex, England, 2004 Search PubMed.
- A. Iqbal, I. Ani and R. Rajput, J. Chem. Technol. Biotechnol., 2012, 87, 177–188 CrossRef CAS PubMed.
- P. Praserthdam, J. Phungphadung and W. Tanakulrungsank, Mater. Res. Innovations, 2003, 7, 118–123 CAS.
- M. H. D. Othman, Z. Wu, N. Droushiotis, U. Doraswami, G. Kelsall and K. Li, J. Membr. Sci., 2010, 351, 196–204 CrossRef CAS PubMed.
- J. D. Nicholas, Low Temperature Constrained Sintering of Cerium Gadolinium Oxide Films for Solid Oxide Fuel Cell Applications, PhD Dissertation, University of California, Berkeley, 2007.
- C. M. Kleinlogel and L. J. Gauckler, J. Electroceram., 2000, 3, 231–243 CrossRef.
- C. M. Kleinlogel and L. J. Gauckler, Solid State Ionics, 2000, 135, 567–573 CrossRef CAS.
- S. Li, C. Xian, K. Yang, C. Sun, Z. Wang and L. Chen, J. Power Sources, 2012, 205, 57–62 CrossRef CAS PubMed.
- W. Zhang, L. T. Kuhn, P. S. Joergensen, B. R. Sudireddy, J. J. Bentzen, C. Bernuy-Lopez, S. Veltze and T. Ramos, J. Power Sources, 2014, 258, 297–304 CrossRef CAS PubMed.
- M. Burbano, S. Nadin, D. Marrocchelli, M. Salanne and G. W. Watson, Phys. Chem. Chem. Phys., 2014, 16, 8320–8331 RSC.
| This journal is © The Royal Society of Chemistry 2015 |