
Polymer coated phosphate glass/hydroxyapatite composite scaffolds for bone tissue engineering applications
 b
and
b
and
Abstract
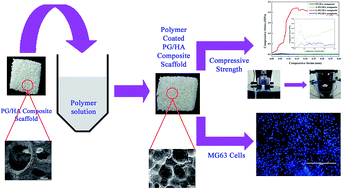
The development of bioactive ceramic composite scaffold materials with enhanced mechanical strength has been a topic of great interest in bone tissue engineering. In the present study, phosphate glass/hydroxyapatite (PG/HA) composite scaffold with an open-porous structure (∼89% porosity and pore size in the range 200–500 μm) has been fabricated by polymer foam replication method. In order to enhance the mechanical strength of this scaffold, a polymer coating of alginate, chitosan and gelatin was made on the composite scaffold by immersion method. The polymer coating did not affect the interconnectivity but reduced the porosity of the PG/HA composite scaffold. Biodegradation studies revealed that all the composite scaffolds have undergone significant degradation. But the compressive strength of the gelatin coated scaffold (G-PG/HA) exhibited sevenfold enhancement than the pristine scaffold. The PG/HA and G-PG/HA composite scaffolds revealed excellent biocompatibility with human osteoblast-like MG-63 cells. Hence, the G-PG/HA composite scaffold may be a smart promising scaffold for bone tissue engineering.
 Please wait while we load your content...
Please wait while we load your content...

