DOI:
10.1039/C5RA09181K
(Paper)
RSC Adv., 2015,
5, 64335-64345
Spectroscopic and molecular docking evidence of aspirin and diflunisal binding to DNA: a comparative study
Received
16th May 2015
, Accepted 13th July 2015
First published on 14th July 2015
Abstract
Aspirin and diflunisal belong to the salicylate class of non-steroidal anti-inflammatory drugs with diverse pharmacological and biological activities. Deciphering the interaction of drugs with DNA not only offers insights for the rational design of novel and more efficient drugs targeted to DNA, but also gives an opportunity for developing effective therapeutic agents for the control of gene expression. A series of spectroscopic studies were performed to ascertain the binding mode of aspirin and diflunisal with calf thymus DNA. UV-visible spectroscopy confirmed aspirin and diflunisal interaction with DNA. Steady state fluorescence experiments revealed a binding constant of 2.3 × 104 L mol−1 for aspirin and 7.9 × 103 L mol−1 for diflunisal. In addition, their binding modes with calf thymus DNA were established by a series of experiments including competitive displacement assays, urea denaturation, iodide quenching, viscosity measurements, DNA melting studies and CD analysis. They corroborated the intercalative binding of aspirin and groove binding of diflunisal with calf thymus DNA. The effect of ionic strength established the role of electrostatic interaction in aspirin–DNA binding processes. Molecular docking studies further complemented the experimental results.
1 Introduction
Studies on molecular interactions between drugs and DNA have become an active area of research in recent years.1,2 DNA is preferred for use as a drug target because of its well-studied three-dimensional structure and the predictability of its accessible chemical and functional groups.3 Regulation of cell functions by targeting DNA via interfering with replication or by modulating transcription seems rational, instinctively appealing and conceptually candid.4 DNA starts transcribing or replicating upon receiving a signal, which is generally in the form of a regulatory proteins binding to a specific region of the DNA. Therefore, if the binding specificity of the regulatory proteins is mimicked by any small molecule then the function of DNA can be modulated artificially.4–6 Thus, these small molecules act as a drug when alteration of DNA function is required to control a disease.5,6 Interaction studies of drug with DNA is not only helpful in understanding the mode of interaction, but also provide great help in designing DNA targeted specific drugs. Many small molecules that bind to DNA are clinically proven therapeutic agents, although their exact mode of action has not been determined. The development of novel molecules that can target specific DNA sequences with high binding affinities can only be accomplished by finding out their molecular recognition patterns.5,6 By understanding the exact mode of action, in vitro designing and screening of new DNA targeted drugs will be possible.5,6
Many small molecules of biological importance are known to interact with DNA involving non-covalent interactions.7,8 Three major modes of non-covalent interactions are electrostatic interactions, groove binding and intercalative binding. Electrostatic binding occurs due to interaction of negatively charged DNA phosphate backbone and positively charged end of small molecules. Two different types of groove binding mode are major groove binding and minor groove binding. Groove binding involves hydrogen bonding or van der Waals interaction with nucleic acid bases. Intercalation occurs when small molecules intercalate within the nucleic acid base pairs.9
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used pharmaceutical drugs. They exhibit favourable anti-inflammatory, analgesic and antipyretic properties and are broadly used for the relief of pain and inflammation.10 Aspirin and diflunisal (Fig. 1), belong to the salicylate class of NSAIDs. NSAIDs, besides having analgesic and anti-inflammatory properties, shows pronounced antitumoral properties by diminishing the number and size of carcinogen-induced colon tumors.11–13 Numerous studies have also revealed that NSAIDs induce the apoptosis of colon, breast, prostate, human myeloid leukaemia and stomach cancer cell lines.14,15 Pharmacological effects of NSAIDs are well established and are mediated by inhibition of enzyme cyclooxygenase.10 Although, the precise molecular mechanism for the antitumor properties of NSAIDs has not been well established. Furthermore, detailed studies of the interaction of aspirin and diflunisal with DNA are not reported from the literature which provided us the impulse to investigate their interaction with DNA in detail. Therefore, the interaction of aspirin and diflunisal with Ct-DNA is worthy of further study and has significant meaning on understanding the binding mode and figuring out the reasons for the difference in biological activities and clinical efficacy of these drugs.
 |
| | Fig. 1 Chemical structure of (a) aspirin (b) diflunisal. | |
Our studies have focussed to evaluate the interaction of aspirin and diflunisal with DNA in vitro by using various biophysical techniques and in silico by exploiting molecular docking. Changes in the absorption and the fluorescence spectra confirmed the formation of complex between drug and DNA. In order to establish the binding mode, several experiments like viscosity measurements, DNA melting studies, CD spectroscopy and molecular docking were employed. Our results clearly demonstrated the intercalation between aspirin and DNA however, groove binding mode was observed between DNA and diflunisal.
2 Experimental
2.1 Materials
Aspirin, diflunisal, calf thymus DNA (Ct-DNA) and Hoechst 33258 were purchased from Sigma Aldrich, USA. Ethidium bromide (EB) was purchased from Himedia, India. All the other chemicals were of reagent grade.
2.2 Sample preparation
Aspirin, diflunisal and Ct-DNA were dissolved in their respective solvents to make stock solution of 10 mM. Working solutions were made according to the requirements. Stock solution of aspirin was prepared in 10% methanol. Diflunisal solution was prepared in methanol and diluted with 0.1 M sodium hydroxide solution. Ct-DNA was dissolved in 10 mM Tris–HCl buffer (pH 7.2) at 4 °C and obtained a homogenous solution with occasional mixing by vortex for 24 h. No further purification of Ct-DNA was done since the absorbance ratio of A260/A280 was between 1.8 and 1.9.16 The concentrations of Ct-DNA solutions were determined by using the average extinction coefficient value of 6600 M−1 cm−1 of a single nucleotide at 260 nm. All reactions were performed in the presence of 10 mM Tris–HCl buffer (pH 7.2) at room temperature.
2.3 UV-visible spectroscopy
In order to study the drug–DNA interaction, UV spectra were recorded with a Beckman DU 40 spectrophotometer (USA) using quartz cuvettes. The UV-spectra of aspirin and aspirin–Ct-DNA complex were recorded in the wavelength range 200–250 nm while, spectra of diflunisal and diflunisal–Ct-DNA complex were recorded in the wavelength range of 200–360 nm. Increasing concentration of Ct-DNA was titrated against 50 μM of aspirin and diflunisal. As the maximum absorption of diflunisal (265 nm) lies close to DNA (260 nm), DNA solutions of same concentrations without drug were used as the blank to observe the UV-spectra specific to drug–DNA complex. The blanks were used for each tube containing same amount of Ct-DNA present in sample without any drug. Thus, after baseline correction using DNA solution as a blank, any measured absorbance is due to the presence of the drug complexed with DNA.
2.4 Fluorescence studies
All fluorescence experiments were carried out by fluorometric titration using a Shimadzu spectrofluorometer-5000 (Japan) equipped with xenon flash lamp using 1.0 cm quartz cells. Excitation was done at 208 nm for aspirin while 265 nm for diflunisal. Emission spectra were recorded from 360 nm to 500 nm for aspirin whereas 330 nm to 540 nm for diflunisal, with the widths of both the excitation and the emission slits set to 5 nm in all fluorescence studies. The fluorescence titration was performed against 50 μM of aspirin and diflunisal with varying concentrations (0–70 μM) of Ct-DNA.
In EB exclusion assay, a solution containing EB (2 μM) and Ct-DNA (20 μM) was titrated with increasing concentration of aspirin and diflunisal. EB–Ct-DNA complex was excited at 475 nm and emission spectra were recorded from 500–700 nm. In another experiment, 2 μM of Hoechst 33258, a well-known groove binder was added to 20 μM of Ct-DNA. Ct-DNA–Hoechst 33258 complex was excited at 343 nm and emission spectra were recorded from 350–600 nm.
Iodide quenching studies were performed in the absence and presence of Ct-DNA. Emission spectra were recorded either in the absence or presence of 50 μM Ct-DNA in 3 mL reaction mixture which included 50 μM aspirin, 10 mM Tris–HCl (pH 7.2) and varying concentration of KI from 0–16 mM. Similar set of titration was done for diflunisal with concentration of KI varying from 0–16 mM.
Urea induced denaturation assay, two cuvettes containing Ct-DNA (50 μM) along with either aspirin (50 μM) or diflunisal (50 μM) were titrated in a total volume of 3 mL by increasing concentration of urea (0–3.60 M). Excitation was done at 208 nm and 265 nm for aspirin and diflunisal respectively while emission spectra were recorded from 330–520 nm.
Effect of ionic strength was studied by varying the concentration of NaCl between 0–40 mM in a total volume of 3 mL using 10 mM Tris–HCl (pH 7.2) containing aspirin (50 μM) or diflunisal (50 μM) and 50 μM Ct-DNA in two different experiments. Excitation was done at 208 nm for aspirin and 265 nm for diflunisal whereas emission spectra were recorded from 350–550 nm.
2.5 Viscosity measurement
Viscosity measurements were performed for further elucidation of the binding mode between aspirin or diflunisal with Ct-DNA. Ct-DNA concentration was kept constant at 100 μM while varying aspirin and diflunisal concentrations. Viscosity measurements were made on an Ubbelohde viscometer which was kept at 25 °C (accuracy ±0.1 °C) by a constant temperature bath. The flow time was measured with a digital stopwatch for three times to get an average calculated time. The data were presented as (η/η0)1/3 versus the ratios of the concentration of aspirin and diflunisal to that of DNA, where η0 and η are the viscosities of DNA in the absence and presence of drug respectively.
2.6 DNA melting studies
DNA melting experiment of Ct-DNA alone, Ct-DNA–aspirin and Ct-DNA–diflunisal complex were performed by monitoring absorbance intensities at different temperatures. The samples contained either Ct-DNA alone (50 μM) or Ct-DNA and aspirin (50 μM each) or Ct-DNA and diflunisal (50 μM each). The absorbance of the samples were monitored at 260 nm and then plotted as a function of temperature ranging from 25 to 100 °C. The values of Tm were determined as the transition midpoint of melting curve.
2.7 Circular dichroism (CD) studies
CD measurements of Ct-DNA, Ct-DNA–aspirin and Ct-DNA–diflunisal were taken on an Applied Photophysics CD spectrophotometer (model CIRASCAN, U.K.) equipped with a Peltier temperature controller to keep the temperature of the sample constant at 25 °C. The molar ratio of DNA concentration to aspirin/diflunisal concentrations was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0, 1:1 and 1:2. All the CD spectra were recorded at wavelengths between 230–300 nm with a scan speed of 200 nm min−1 and a spectral band width of 10 nm. Average of three scans was taken in all the experiments. Background spectrum of buffer solution (10 mM Tris–HCl, pH 7.2) was subtracted from the spectra of Ct-DNA and Ct-DNA–aspirin, Ct-DNA–diflunisal complex.
:0, 1:1 and 1:2. All the CD spectra were recorded at wavelengths between 230–300 nm with a scan speed of 200 nm min−1 and a spectral band width of 10 nm. Average of three scans was taken in all the experiments. Background spectrum of buffer solution (10 mM Tris–HCl, pH 7.2) was subtracted from the spectra of Ct-DNA and Ct-DNA–aspirin, Ct-DNA–diflunisal complex.
2.8 Molecular docking
Molecular docking studies were performed using HEX 8.0.0 software, an interactive molecular graphic program.16 The crystal structure of B-DNA dodecamer d(CGCGAATTCGCG)2 (PDB ID: 1BNA) was downloaded from the protein data bank (http://www.rcsb.org./pdb). Mol file of aspirin and diflunisal was obtained from https://www.pubchem.ncbi.nlm.nih.gov/ and further converted into PDB format using Avogadro's 1.01.17 The Hex 8.0.0 performs docking using Spherical Polar Fourier Correlations. It necessitates the ligand and the receptor as input in PDB format. The parameters that were used for docking include: correlation type – shape only, FFT mode – 3D, grid dimension – 0.6, receptor range – 180, ligand range – 180, twist range – 360, distance range – 40. The docking poses are then visualized by using PyMol software (DeLano Scientific, San Carlos, CA, USA).
3 Result and discussion
3.1 Interaction of aspirin and diflunisal with DNA
3.1.1 UV-visible spectroscopy. UV-visible spectroscopy is a very simple method which helps in depicting the structural changes and also sheds light on the complex formation.18,19 The absorption spectra of aspirin and diflunisal obtained in the absence and presence of Ct-DNA are shown in (Fig. 2). In the UV region, aspirin has absorption peaks (Fig. 2a) at 208 nm and 225 nm (ref. 20) while diflunisal displays a peak (Fig. 2b) at 265 nm.21 On addition of Ct-DNA, hyperchromism was observed without any noticeable shift in the position of maximum absorption peak that clearly indicated the formation of complex between Ct-DNA and aspirin/diflunisal.16,22,23 However, absence of any clear isobestic point implies that 1:1 drug:DNA stoichiometry is not maintained during the binding process and there is more than one type of binding which further prevented the calculation of binding constant.24 The exact binding mode cannot be established simply by this technique, hence further experiments were required to explore the mode of interaction.
 |
| | Fig. 2 UV-visible absorption spectra of aspirin and diflunisal (50 μM) in presence of increasing concentrations of Ct-DNA in Tris–HCl buffer (pH 7.2). The arrows show the changes upon increasing amounts of Ct-DNA. | |
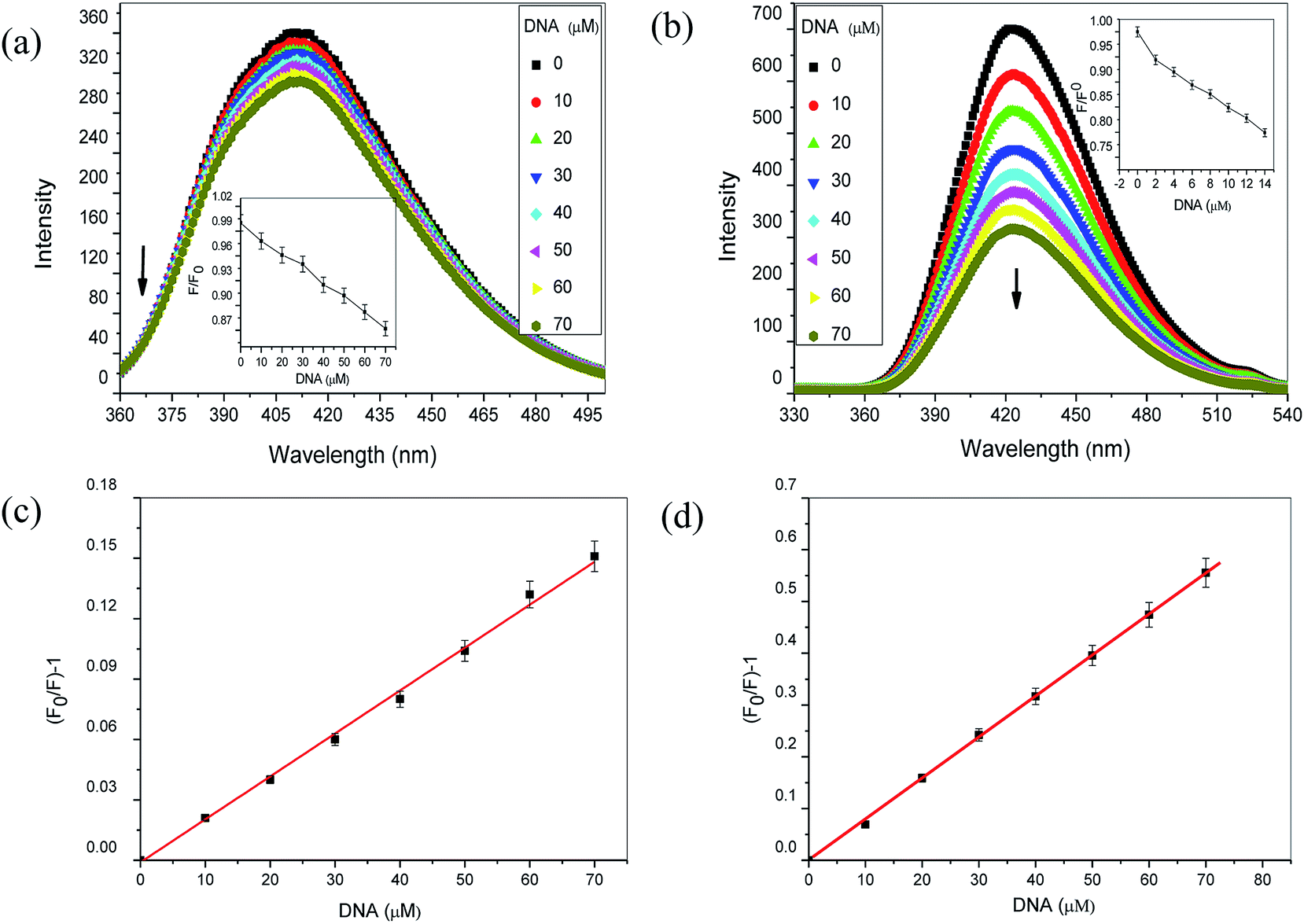
3.1.2 Steady state fluorescence. To further explore the interaction of aspirin and diflunisal with Ct-DNA, steady state fluorescence was performed. As the endogenous fluorescence of Ct-DNA is very weak, we used drugs (aspirin and diflunisal) as a fluorescence probe to examine the interaction between DNA and drugs. Fluorescence quenching studies were performed by maintaining the fixed concentration of drugs (50 μM) while varying the concentration of Ct-DNA. Aspirin exhibited an emission spectrum at 415 nm when excited at 208 nm (Fig. 3a) while diflunisal showed a peak with maxima around at 422 nm when excited at 265 nm (Fig. 3b). With increasing addition of Ct-DNA, the intensity at 415 nm and 422 nm peaks fluorescence decreased gradually for aspirin and diflunisal respectively which strengthen the interaction between Ct-DNA and drugs. The inset in Fig. 3a and b shows that within the examined concentrations range decreased fluorescence intensity of aspirin and diflunisal was directly proportional to the Ct-DNA concentration. Further, Stern–Volmer quenching constant (KSV) was calculated (Fig. 3c and d) by using following well known Stern–Volmer equation.where F0 and F represents the fluorescence intensities in the absence and presence of the quencher Ct-DNA [Q] respectively and KSV is the Stern–Volmer quenching constant, which is a measure of the efficiency of quenching by DNA. The slopes of the (F0/F) vs. [Ct-DNA] plots yield the values of KSV. In case of aspirin it was calculated to be 2.3 × 104 L mol−1, which was found to be coherent with intercalative binding mode.25 Therefore, binding mode of aspirin with Ct-DNA is most likely to be intercalative. Again, KSV value for diflunisal was obtained to be 7.9 × 103 L mol−1, which was much lower than the intercalators25 and hence clearly indicated that diflunisal binds with Ct-DNA by non-intercalative binding mode.
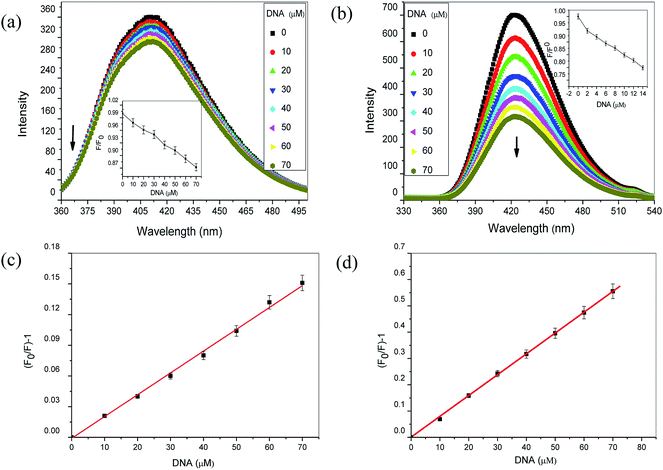 |
| | Fig. 3 Fluorescence emission spectra of (a) aspirin (50 μM) and (b) diflunisal (50 μM) in the presence of increasing concentrations of Ct-DNA. Insets show the variation of the respective fluorescence intensities with DNA concentrations. Arrow shows the decrease in intensity upon increasing the concentration of Ct-DNA. Data represent mean ± SD of three different experiments. Stern–Volmer plot of (c) aspirin and (d) diflunisal with increasing concentration of Ct-DNA. Stern–Volmer quenching constants were calculated to be 2.3 × 104 L mol−1 and 7.9 × 103 L mol−1 for aspirin and diflunisal respectively. | |
Linear Stern–Volmer plots were obtained for both aspirin and diflunisal, suggesting that only one type of the quenching process occurred, either static or dynamic quenching.26 Process of quenching was further confirmed by calculating the values of biomolecular quenching rate constants (Kq), which are evaluated by using the following equation
where
τ0 is the average lifetime of molecule without quencher. Since fluorescence lifetimes are typically near 10
−8 s,
Kq was calculated from the above equation and was found to be of the order of 2.3 × 10
12 L mol
−1 s
−1 for aspirin and 7.9 × 10
11 L mol
−1 s
−1 for diflunisal. The
Kq values of aspirin and diflunisal were greater than the value of the maximum scatter collision quenching constant (2.0 × 10
10 L mol
−1 s
−1),
26 which depicts that probable quenching mechanism of aspirin and diflunisal with DNA, was initiated by complex formation rather than by dynamic collision. The values of
KSV and
Kq are listed in
Table 1.
Table 1 Various parameters for aspirin and diflunisal interaction with DNA
| Drug |
KSV (L mol−1) |
Kq (L mol−1 s−1) |
Ra |
| R is the correlation coefficient. |
| Aspirin |
2.3 × 104 |
2.3 × 1012 |
0.9964 |
| Diflunisal |
7.9 × 103 |
7.9 × 1011 |
0.9966 |
3.2 Binding mode of aspirin and diflunisal with DNA
3.2.1 Competitive displacement assay. To further investigate the binding mode of aspirin and diflunisal with Ct-DNA, the ethidium bromide (EB) displacement assay was carried out. EB is a planar aromatic fluorescent dye which does not show any appreciable emission in the aqueous solution, however in the presence of DNA it is strongly emissive due to its intercalation within the DNA base pairs.27 Since EB intercalates DNA, competitive displacement of EB on addition of the drug will be suggestive of an intercalative binding mode. Fig. 4a and b shows the emission spectra of the Ct-DNA–EB system due to aspirin and diflunisal respectively. With increasing concentration of aspirin, a remarkable decrease in fluorescence of Ct-DNA–EB system was observed (Fig. 4a). This suggested that aspirin substituted EB in the Ct-DNA–EB system and dissociated the EB into the solvent. This lead to a decrease in the fluorescence intensity of the Ct-DNA–EB system and hence provided a direct evidence in support of intercalative binding mode.
 |
| | Fig. 4 Fluorescence titration of Ct-DNA–EB complex with aspirin and diflunisal. Ct-DNA (20 μM) was dissolved in 10 mM Tris–HCl (pH 7.2) and EB was added to a final concentration of 2 μM. Fluorescence quenching studies were performed with varying concentration of (a) aspirin and (b) diflunisal. EB–Ct-DNA complex was excited at 475 nm and emission spectra were recorded from 500–700 nm. Fluorescence titration of Ct-DNA (20 μM) and (2 μM) Hoechst (groove binder) complex with (c) aspirin and (d) diflunisal. Ct-DNA–Hoechst complex was excited at 343 nm and emission spectra were recorded from 350–600 nm. | |
In case of diflunisal there was no significant change in the fluorescence intensity (Fig. 4b). This clearly indicated that diflunisal does not replace EB from Ct-DNA helix and thus binds to Ct-DNA primarily via non-intercalative mode. To further support our finding we used Hoechst 33258, which binds to the minor groove of double stranded B-DNA. Hoechst 33258–DNA complex gives a characteristic emission peak at around 454 nm when excited at 343 nm. The emission spectra is due to the binding of Hoechst 33258 to the minor groove of DNA.28,29 On addition of aspirin, there was no change in the fluorescence intensity of the Ct-DNA–Hoechst system (Fig. 4c) which ruled out the minor groove binding and further confirmed the intercalative binding mode of aspirin with Ct-DNA. Furthermore, it is apparent from the (Fig. 4d) that the increasing addition of diflunisal results in a substantial reduction of the fluorescence intensity of Ct-DNA–Hoechst system at 454 nm indicating that diflunisal replaced Hoechst 33258 from the minor groove of DNA, ruling out the intercalative binding.
These results indicate that aspirin and diflunisal follow intercalative binding and groove binding mode with Ct-DNA respectively.
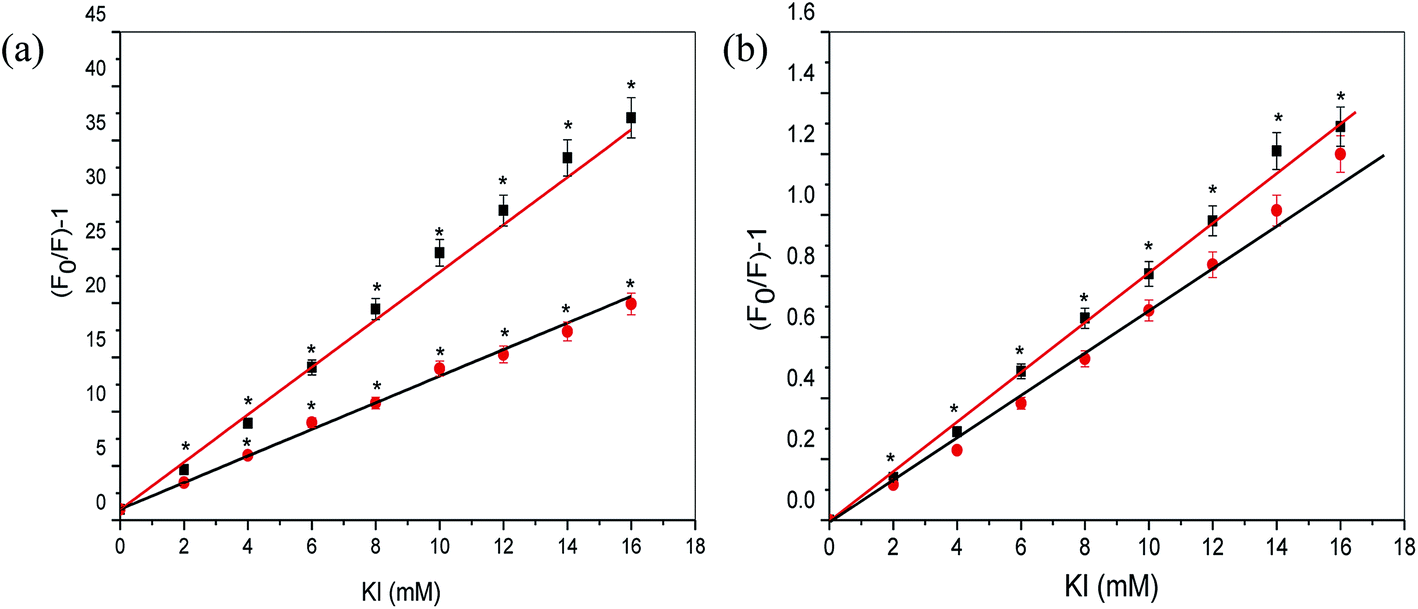
3.2.2 KI quenching studies. Another reliable method for investigating aspirin/diflunisal binding mode with Ct-DNA is the iodide quenching experiments. In this study, negatively charged KI was taken as it can effectively quench the fluorescence intensity of small molecules. Approach of any anionic quencher towards DNA molecule is restricted due to the repulsion from the negatively charged phosphate groups on DNA backbone. Hence, small molecules intercalated into the DNA helix are well protected. However, molecules bound to the surface/groove of DNA are easily accessible to the quencher as they are much exposed to the surrounding environment.30,31 Fig. 5 shows the Stern–Volmer plots for KI quenching of aspirin and diflunisal fluorescence in the absence and presence of Ct-DNA. KSV values were calculated by using the eqn (1). The calculated KSV values for aspirin–KI and aspirin–KI–Ct-DNA were found to be 22.91 and 11.17 L mol−1 respectively. It is apparent from KSV values that the KI could effectively quench the fluorescence of aspirin in a buffer solution as compared to aspirin–Ct-DNA complex (Fig. 5a). Therefore, it could be established that the probable interaction mode of aspirin with Ct-DNA might be intercalative binding. On the other hand, KSV values for diflunisal–KI and diflunisal–KI–Ct-DNA were found to be 8.4 and 7.2 L mol−1. It is clearly evident from KSV values that in buffer solution iodide quenched the fluorescence of diflunisal efficiently. However the KSV values of diflunisal–KI in presence of Ct-DNA, decreased slightly which indicated that diflunisal could be partly protected (Fig. 5b). Thus, it could be confirmed that groove binding mode of interaction occurs between diflunisal and Ct-DNA. Table 2 summarizes various parameters obtained in KI quenching studies with the help of Stern–Volmer plots.
 |
| | Fig. 5 KI quenching studies. Stern–Volmer plot for fluorescence quenching of (a) aspirin (50 μM) and (b) diflunisal (50 μM) by KI in the absence and presence of Ct-DNA (50 μM) in 10 mM Tris–HCl buffer (pH 7.2). Concentration of KI was varied from 0 to 16 mM. Data represent mean ± SD of three experiments. *p value < 0.05 as compared to control. | |
Table 2 Parameters obtained in KI quenching studies in absence and presence of Ct-DNA environments
| Drug |
Aspirin |
Ra |
S.D.b |
Diflunisal |
Ra |
S.D.b |
| R is the correlation coefficient. S.D. is standard deviation. |
| KSV (L mol−1) in buffer |
22.91 ± 0.91 |
0.9988 |
0.8612 |
8.4 ± 0.41 |
0.9983 |
0.7904 |
| KSV (L mol−1) in drug–DNA complex |
11.17 ± 0.53 |
0.9983 |
0.7424 |
7.2 ± 0.31 |
0.9975 |
0.7125 |
| Relative reduction in KSV (%) |
51 |
|
|
14 |
|
|
3.2.3 Effect of urea on DNA binding. Urea is a well-known denaturant which destabilizes the double stranded DNA helix resulting in the release of entrapped drug molecules, leading to a change in the emission spectrum of the drug molecules. Therefore, denaturation experiment due to urea is exploited to study the binding mode of small molecules.30,32 As evident from (Fig. 6a), with the addition of urea to the aspirin bound Ct-DNA leads to a progressive increase in the fluorescence intensity of soluble aspirin due to the destabilisation of DNA helix in presence of urea. This increase in the fluorescence intensity of aspirin–Ct-DNA complex is probably due to the release of aspirin molecule entrapped in DNA helix. This clearly demonstrated the proposed intercalative binding mode of aspirin with Ct-DNA. Moreover, the fluorescence intensity of diflunisal with increasing concentration of urea remains unchanged (Fig. 6b). No change in the fluorescence intensity of diflunisal was observed even after the addition of urea upto 3.60 M which clearly indicated that diflunisal binds to Ct-DNA via non-intercalative binding. The inset in Fig. 6a and b depicts the relative extent of binding of aspirin and diflunisal. Ratio of peak fluorescence intensities in the presence and absence of urea (F/F0) has been plotted as a function of urea concentration.
 |
| | Fig. 6 Fluorescence spectra of aspirin/diflunisal–Ct-DNA complex with urea. Fluorescence intensity of Ct-DNA bound (a) aspirin and (b) diflunisal with addition of urea. Aspirin–Ct-DNA system was excited at 208 nm and diflunisal–Ct-DNA system was excited at 265 nm while emission spectra were recorded from 330–520 nm. Inset shows the variation of fluorescence intensity of Ct-DNA bound aspirin/diflunisal, as a function of urea concentration. Data represent mean ± SD of three experiments. *p value < 0.05 as compared to control. | |
3.2.4 Viscosity measurement. Viscosity measurements are regarded as the most consistent method for elucidating the binding mode of small molecules with DNA as they are sensitive to change in DNA length upon addition of a ligand. In case of classical intercalation, binding of a ligand lengthen the DNA helix as base pairs are separated to accommodate such ligand. This results in increased viscosity of DNA solution. In contrast, a ligand does not cause any obvious increase in the viscosity of DNA solution if bound to grooves.18,33,34 A plot of (η/η0)1/3 versus [aspirin or diflunisal]/[DNA] gives a measure of the viscosity changes. The viscosity of Ct-DNA increased remarkably with increasing the concentrations of aspirin (Fig. 7a) and thus clearly established that intercalation binding mode existed between aspirin and Ct-DNA. However, it is apparent that the addition of diflunisal had no obvious effect on the relative viscosity of Ct-DNA (Fig. 7b), indicating that the interaction is a groove binding, not an intercalative binding mode.
 |
| | Fig. 7 Effect of increasing concentration of (a) aspirin and (b) diflunisal on the viscosity of 100 μM Ct-DNA solution. Data represent mean ± SD of three determinations. | |
3.2.5 DNA melting studies. Another reliable method to investigate the binding of aspirin and diflunisal to DNA was obtained by thermal denaturation experiment. At certain temperature, local openings of the DNA double helix extend over the entire molecule and subsequently complete the separation of two strands.32,35 Intercalation of small molecules within the double helix is known to increase the DNA melting temperature (Tm) by about 5–8 °C, owing to the increased stability of the helix in the presence of an intercalator while the groove binding and electrostatic binding causes no obvious increase in Tm.36,37 Tm values was determined by monitoring the absorbance of the DNA bases at 260 nm as a function of temperature ranging from 25 °C to 100 °C. The melting curves of Ct-DNA in the absence and presence of aspirin and diflunisal are presented in Fig. 8. For each observed transition, Tm was determined from the midpoint of the melting curves. As evident from Fig. 8a, with increasing concentration of aspirin, Tm of Ct-DNA was increased from 66.8 ± 1 °C to 72.4 ± 1 °C, due to increased DNA stability in presence of aspirin. These results are strongly suggestive of intercalation of aspirin into the Ct-DNA double helix. On the other hand, in presence of diflunisal the observed Tm value of Ct-DNA was 68.5 ± 1 °C (Fig. 8b). This clearly ruled out the intercalation of diflunisal and further strengthens the possibility for groove binding to the Ct-DNA.
 |
| | Fig. 8 Thermal melting profile of Ct-DNA (50 μM) in the absence (■) and presence (●) of (a) aspirin and (b) diflunisal. Absorbance was taken at 260 nm. | |
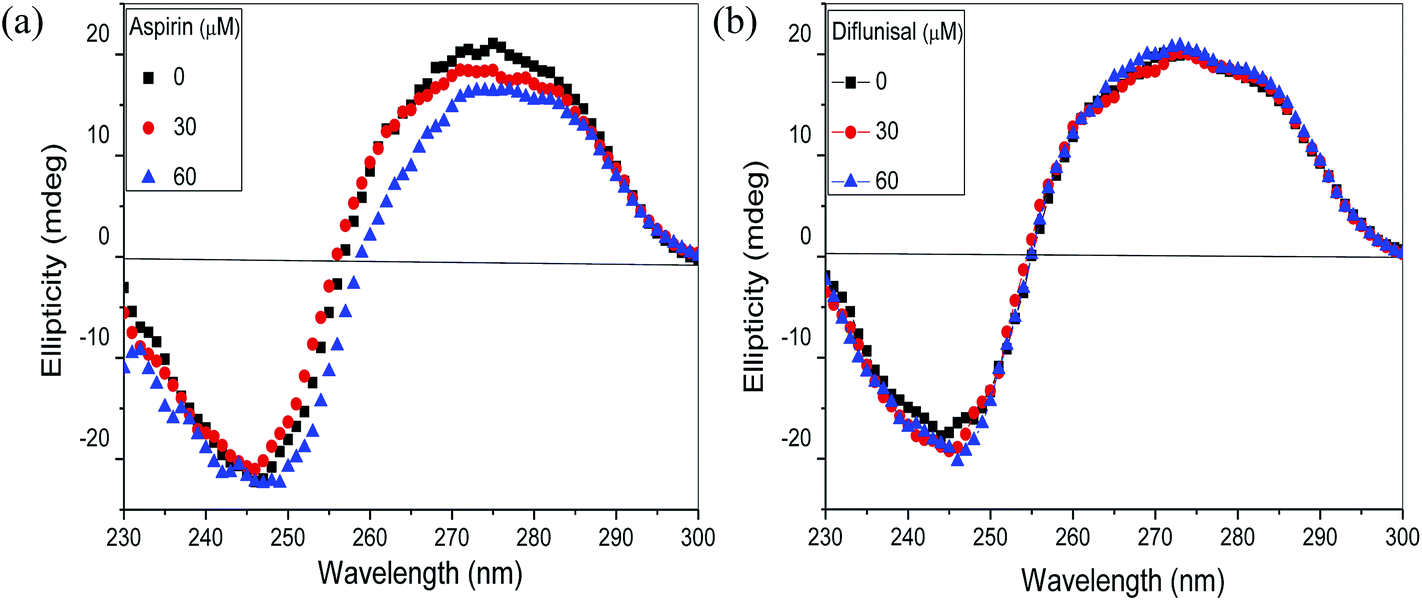
3.2.6 Circular dichroism studies. CD is an extremely sensitive method of studying any conformational alteration in the DNA backbone. This technique is useful to examine non-covalent DNA–ligand interactions.38,39 As depicted in (Fig. 9) the CD spectrum of free DNA solution exhibited a typical B-form conformation with a positive peak at around 275 nm corresponding to base pair stacking and a negative band at around 244 nm due to the helicity. It has been reported that an intercalator changes the intensities of both the bands whereas no obvious perturbation of the base stacking and helicity bands occurs in case of groove binding and electrostatic interaction.28,40 As apparent from (Fig. 9a), ellipticity of both the bands were changed with subsequent addition of aspirin. Similar changes in peak at 244 nm were observed in presence of both aspirin and diflunisal, which occurs due to the alteration in the hydration layer of DNA. However, change in the intensity of the CD peak at 275 nm was observed only in case of aspirin. Such change could be due to the disruption of the stacked nitrogenous bases in order to accommodate the aspirin within the DNA helix. Thus, we confirmed our findings that aspirin binds to DNA in an intercalative manner. Negligible change in the CD spectra of DNA was observed with increasing concentration of diflunisal (Fig. 9b), confirming the groove binding mode of diflunisal with Ct-DNA.
 |
| | Fig. 9 CD spectra of Ct-DNA (30 μM) in 10 mM Tris–HCl (pH 7.2) with varying concentration of (a) aspirin and (b) diflunisal. Each spectrum was obtained at 25 °C with a 10 mm path length cell. | |
3.2.7 Molecular docking. In order to establish the binding mode of drugs with DNA, in silico molecular docking was employed. It is an attractive tool for obtaining information about the structural features of ligand–receptor complexes and the binding affinity of a ligand with its receptor, which can corroborate the experimental results.41 In this context, docking studies were performed in an attempt to ascertain the interaction mode of aspirin and diflunisal with DNA. In our experiments, rigid molecular docking were performed to decipher the binding mode of aspirin and diflunisal with a DNA duplex of sequence d(CGCGAATTCGCG)2 dodecamer (PDBID: 1BNA). The ligand has been made flexible to attain different conformations in order to predict the best ligand–receptor fit, and the best energetically favourable docked pose was analysed. It is apparent from Fig. 10a–d, that aspirin interacts with DNA via intercalative binding mode. The resulting relative binding energy of docked DNA–aspirin complex was found to be −4.71 kcal mol−1. Further analysis of docking results revealed that aspirin binds to a GC rich region (Fig. 10a). It has been reported that an intercalator preferably bind to GC rich sequence while groove binders, preferably bind to AT rich sequence.42,43 Furthermore, there is possibility of hydrogen bonding as the oxygen-bearing groups of aspirin (O1) are in proximity to the deoxyguanosine (DG10 of chain A) of the dodecamer Fig. 10b–d. Distance between the hydrogen bonds was found to be 3.2 Å and 3.5 Å (Fig. 10d). It is obvious from Fig. 10e that diflunisal fits snugly into the curved contour of the B-DNA in the minor groove and therefore clearly revealed the groove binding mode of diflunisal with DNA. The binding energy of the DNA–diflunisal complex system was −3.65 kcal mol−1. Furthermore, docking results clearly shown the formation of a hydrogen bond between the oxygen bearing group (O1) of diflunisal and number eight thymidine of one strand of DNA (B chain of DNA) (Fig. 10f and g), with the oxygen (O2) atom serving as a hydrogen bond receptor. As seen in Fig. 10h, distance between the hydrogen bond was found to be 2.1 Å. At pH 7.2 both aspirin and diflunisal carry a net negative charge (http://www.chemicalize.org/structure). Regardless of the electrostatic repulsion of DNA with aspirin and diflunisal, large negative value of the binding energy indicated a higher binding potential of aspirin and diflunisal with DNA. Finally, we confirmed that there is a mutual complement between spectroscopic techniques and molecular docking, which provides valuable information about the binding mode of aspirin and diflunisal with DNA.
 |
| | Fig. 10 Molecular docked structure of aspirin (a–d) and diflunisal (e–h) complexed with DNA showing (a) the binding of aspirin to GC region of dodecamer duplex of sequence [(CGCGAATTCGCG)2 (PDB ID: 1BNA)], (b–d) the possibility of hydrogen bonding (O⋯H: 3.2 Å and O⋯H: 3.5 Å) between the oxygen bearing group (O1) of aspirin and tenth guanine of A chain, (e) binding of diflunisal to the minor groove, (f and g) the formation of a hydrogen between the oxygen bearing group (O1) of diflunisal and number eight thymidine of B chain of DNA and (h) showing the distance of hydrogen bond between diflunisal and B chain (O⋯H: 2.1 Å). The relative binding energies was found to be −4.71 kcal mol−1 for aspirin–Ct-DNA system while for diflunisal–Ct-DNA system it was −3.65 kcal mol−1. | |
3.3 Role of electrostatic interaction
A series of experiments confirmed the binding mode of drugs with Ct-DNA involving hydrogen bonds and electrostatic interactions. In order to confirm the involvement of electrostatic interaction between the drugs and Ct-DNA, the effect of ionic strength was studied. Increased ionic strength screens the negatively charged phosphate backbone of DNA and weakens the interaction between Ct-DNA and drugs due to competition for phosphates.33,44 It is apparent that intercalative binding and groove binding molecule affect with the groove in the DNA double helix, but the electrostatic binding takes place out of the groove. In our experiments, the fluorescence of aspirin–Ct-DNA and diflunisal–Ct-DNA system with increasing concentration of NaCl was studied. Gradual addition of NaCl to aspirin–DNA complex increased the fluorescence intensity (Fig. 11a). This observation could be explained on the basis that with increasing concentration of NaCl, negatively charged DNA phosphate backbone is neutralised leading to contraction of the DNA helix. This results in the release of aspirin in buffer solution enhancing the fluorescence intensity as observed in (Fig. 11a). Accordingly, electrostatic interaction might be involved in the aspirin–Ct-DNA binding process. On the other hand, continuous addition of NaCl to diflunisal–DNA complex showed no significant change in the fluorescence intensity (Fig. 11b). Thus, electrostatic interaction may have negligible role in diflunisal–Ct-DNA interaction. The relative extent of fluorescence intensity of Ct-DNA bound aspirin and diflunisal was given as a function of NaCl concentration, is depicted in inset (Fig. 11a and b).
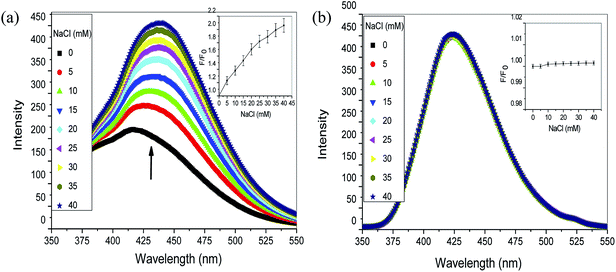 |
| | Fig. 11 Effect of ionic strength on the fluorescence intensity of (a) aspirin–Ct-DNA complex and (b) diflunisal–Ct-DNA. Inset shows the variation of fluorescence intensity of Ct-DNA bound aspirin and diflunisal, as a function of NaCl concentration. Excitation was done at 208 nm for aspirin and 265 nm for diflunisal whereas emission spectra were recorded from 350–550 nm. | |
4 Conclusion
In summary, the present study reports the binding interaction of aspirin and diflunisal with Ct-DNA. The detailed UV-visible absorption spectra and fluorescence spectra studies undertaken in the present work are in total agreement with the intercalative binding mode of aspirin with DNA. Electrostatic interaction might be involved in aspirin–Ct-DNA binding process. Finally, viscosity measurements, DNA melting studies and CD studies further confirmed the intercalation as the most possible binding mode of aspirin with DNA. The in silico molecular docking revealed the binding of aspirin within the GC base pairs of DNA and provided the visual representation of the intercalative binding mode. The interactive mode of diflunisal with Ct-DNA has been established to be groove binding by performing a series of experiments mentioned above. Our results confirmed the prospective probability of using aspirin and diflunisal as an efficient intercalative and groove binding DNA probe, respectively, and also open up the use of these drugs to further development of salicylate based pharmacologically important molecules.
Conflict of interest
The authors declare that there is no conflict of interest in this work.
Acknowledgements
Authors are thankful to UGC, New Delhi, for the award of UGC-MANF-SRF to MAH & TS & CSIR-SRF to SUR & HMI. A research project (Grant No. BT/PR8032/BID/7/443/2013) funded by Department of Biotechnology, New Delhi is thankfully acknowledged. We are also thankful to the Department of Biochemistry A.M.U., Aligarh for providing us the necessary facilities. We also thank the Advanced Instrumentation Research Facility, Jawaharlal Nehru University, New Delhi, for carrying out CD experiments.
References
- J. Li, S. Shuang and C. Dong, Talanta, 2009, 77, 1043–1049 CrossRef CAS PubMed.
- S. Rauf, J. J. Gooding, K. Akhtar, M. A. Ghauri, M. Rahman, M. A. Anwar and A. M. J. Khalid, J. Pharm. Biomed. Anal., 2005, 37, 205–217 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- Y. Shi, C. Guo, Y. Sun, Z. Liu, F. Xu, Y. Zhang, Z. Wen and Z. Li, Biomacromolecules, 2011, 12, 797–803 CrossRef CAS PubMed.
- T. Mavromoustakos, S. Durdagi, C. Koukoulitsa, M. Simcic, M. G. Papadopoulos, M. Hodoscek and S. G. Grdadolnik, Curr. Med. Chem., 2011, 18, 2517–2530 CrossRef CAS.
- B. A. D. Neto and A. A. M. Lapis, Molecule, 2009, 14, 1725–1746 CrossRef CAS PubMed.
- A. Rescifina, C. Zagni, M. G. Varrica, V. Pistarà and A. Corsaro, Eur. J. Med. Chem., 2014, 74, 95–115 CrossRef CAS PubMed.
- W. Hu, S. Deng, J. Huang, Y. Lu, X. Le and W. Zheng, J. Inorg. Biochem., 2013, 127, 90–108 CrossRef CAS PubMed.
- W. D. Sasikala and A. Mukherjee, J. Phys. Chem. B, 2012, 116, 12208–12212 CrossRef CAS PubMed.
- C. E. Dugowson and P. Gnanashanmugam, Phys. Med. Rehabil. Clin., 2006, 17, 347–354 CrossRef PubMed.
- D. J. A. de Groot, E. G. E. de Vries, H. J. M. Groen and S. de Jong, Crit. Rev. Oncol. Hematol., 2007, 61, 52–69 CrossRef CAS PubMed.
- J. I. Johnsen, M. Lindskog, F. Ponthan, I. Pettersen, L. Elfman, A. Orrego, B. Sveinbjornsson and P. Kogner, Cancer Lett., 2005, 228, 195–201 CrossRef CAS PubMed.
- K. Kim, J. Yoon, J. K. Kim, S. J. Baek, T. E. Eling, W. J. Lee, J. Ryu, J. G. Lee, J. Lee and J. Yoo, Biochem. Biophys. Res. Commun., 2004, 325, 1298–1303 CrossRef CAS PubMed.
- D. H. Woo, I. S. Han and G. Jung, Life Sci., 2004, 75, 2439–2449 CrossRef CAS PubMed.
- L. Klampfer, J. Cammenga, H. G. Wisniewski and S. D. Nimer, Blood, 1999, 93, 2386–2394 CAS.
- M. A. Husain, T. Sarwar, S. U. Rehman, H. M. Ishqi and M. Tabish, Phys. Chem. Chem. Phys., 2015, 17, 13837–13850 RSC.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- X. Zhou, G. Zhang and J. Pan, Int. J. Biol. Macromol., 2015, 74, 185–194 CrossRef CAS PubMed.
- H. Guo, C. Cai, H. Gong and X. Chen, Spectrochim. Acta, Part A, 2011, 79, 92–96 CrossRef CAS PubMed.
- S. Z. Bathaie, L. Nikfarjam, R. Rahmanpour and A. A. Moosavi-Movahedi, Spectrochim. Acta, Part A, 2010, 77, 1077–1083 CrossRef CAS PubMed.
- M. E. Abdel-Hamid, N. M. Najib, M. S. Suleiman and Y. M. el-Sayed, Analyst, 1987, 112, 1527–1530 RSC.
- K. P. Rakesh, P. K. Shiva and P. K. Shridhara, Int. J. Res. Chem. Environ., 2012, 2, 221–225 CAS.
- N. Shahabadi, S. Kashanian, M. Mahdavi and N. Sourinejad, Bioinorg. Chem. Appl., 2011, 2011, 525794 Search PubMed.
- D. Sarkar, P. Das, S. Basak and N. J. Chattopadhyay, J. Phys. Chem. B, 2008, 112, 9243–9249 CrossRef CAS PubMed.
- Y. Cao and X. W. He, Spectrochim. Acta, Part A, 1998, 54, 883–892 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Berlin, 3rd edn, 2006, pp. 278–282 Search PubMed.
- Y. Ma, G. Zhang and J. Pan, J. Agric. Food Chem., 2012, 60, 10867–10875 CrossRef CAS PubMed.
- S. U. Rehman, T. Sarwar, H. M. Ishqi, M. A. Husain, Z. Hasan and M. Tabish, Arch. Biochem. Biophys., 2015, 566, 7–14 CrossRef CAS PubMed.
- R. Sarkar and S. K. Pal, Biomacromolecules, 2007, 8, 3332–3339 CrossRef CAS PubMed.
- X. L. Li, Y. J. Hu, H. Wang, B. Q. Yu and H. L. Yue, Biomacromolecules, 2012, 13, 873–880 CrossRef CAS PubMed.
- D. Sahoo, P. Bhattacharya and S. Chakravorti, J. Phys. Chem. B, 2010, 114, 2044–2050 CrossRef CAS PubMed.
- M. A. Husain, Z. Yaseen, S. U. Rehman, T. Sarwar and M. Tabish, FEBS J., 2013, 280, 6569–6580 CrossRef CAS PubMed.
- S. U. Rehman, Z. Yaseen, M. A. Husain, T. Sarwar, H. M. Ishqi and M. Tabish, PLoS One, 2014, 9, e93913 Search PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS.
- A. Wildes, N. Theodorakopoulos, J. Valle-Orero, S. Cuesta-López, J. L. Garden and M. Peyrard, Phys. Rev. Lett., 2011, 106, 048101 CrossRef.
- T. Sarwar, M. A. Husain, S. U. Rehman, H. M. Ishqi and M. Tabish, Mol. BioSyst., 2015, 11, 522–531 RSC.
- S. Bi, H. Zhang, C. Qiao, Y. Sun and C. Liu, Spectrochim. Acta, Part A, 2008, 69, 123–129 CrossRef PubMed.
- T. Sarwar, S. U. Rehman, M. A. Husain, H. M. Ishqi and M. Tabish, Int. J. Biol. Macromol., 2015, 73, 9–16 CrossRef CAS PubMed.
- C. N. N'soukpoé-Kossi, A. A. Ouameur, T. Thomas, A. Shirahata, T. J. Thomas and H. A. Tajmir-Riahi, Biomacromolecules, 2008, 9, 2712–2718 CrossRef PubMed.
- N. Grover, N. Gupta, P. Singh and H. H. Thorp, Inorg. Chem., 1992, 31, 2014–2020 CrossRef CAS.
- G. Macindoe, L. Mavridis, V. Venkatraman, M. D. Devignes and D. W. Ritchie, Nucleic Acids Res., 2010, 38, W445–W449 CrossRef CAS PubMed.
- S. U. Rehman, T. Sarwar, M. A. Husain, H. M. Ishqi and M. Tabish, Arch. Biochem. Biophys., 2015, 576, 49–60 CrossRef PubMed.
- J. Ren and J. B. Chaires, Biochemistry, 1999, 38, 16067–16075 CrossRef CAS PubMed.
- F. Arjmand and A. Jamsheera, Spectrochim. Acta, Part A, 2011, 78, 45–51 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.