
Structure–property relationship of isomeric diphenylethenyl-disubstituted dimethoxycarbazoles†
Abstract
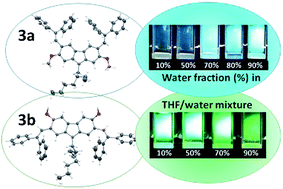
Isomeric 3,6-dimethoxy- and 2,7-dimethoxycarbazoles containing diphenylethenyl moieties were synthesized by condensation of the appropriate dimethoxycarbazoles with diphenylacetaldehyde. The solid-state structures and the molecular order of the compounds were proven by X-ray crystallography. Both compounds were found to be capable of glass formation with comparable glass transition temperatures (70–71 °C). They exhibited high thermal stabilities, with the 5% weight loss temperatures exceeding 375 °C. The isomer having diphenylethenyl groups at C-3 and C-6 positions and methoxy groups at C-2 and C-7 positions (3a) exhibited aggregation-induced emission (AIE), while its counterpart having diphenylethenyl groups at C-2 and C-7 positions and methoxy groups at C-3 and C-6 positions (3b) showed the opposite effect, i.e. aggregation-caused quenching (ACQ). The derivative 3b showed superior charge transporting properties. Time-of-flight hole drift mobilities in its layers approached 10−3 cm2 V−1 s−1 at high electric fields. A comparative theoretical analysis of the compounds was performed using density functional theory (DFT) and time-dependent DFT calculations. They proved more effective π-conjugation in the derivative of 3,6-dimethoxy carbazole (3b), which was also observed by UV and fluorescence spectroscopies. The theoretical study revealed relatively low ground state dipole moment of 0.69 D of the isomer 3b, while its counterpart (3a) showed much higher ground state dipole moment of 5.98 D. The difference in polarity was found to have the crucial effect on the molecular arrangement in the crystals and consequently, on the thermal transitions and charge-transporting properties.
 Please wait while we load your content...
Please wait while we load your content...

