
The one-step oxidation of methanol to dimethoxymethane over sulfated vanadia–titania catalysts: influence of calcination temperature
Heqin Guo,
Debao Li,
Congbiao Chen,
Litao Jia and
Bo Hou*
State Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China. E-mail: houbo@sxicc.ac.cn; Fax: +86 351 4040087; Tel: +86 351 4040087
First published on 20th July 2015
Abstract
Sulfated vanadia–titania catalysts were prepared by the rapid combustion method and calcined at different temperatures. The influence of calcination temperature on the physicochemical properties of the catalysts was characterized by nitrogen adsorption (BET), X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), inductively coupled plasma-optical emission spectroscopy (ICP-OES), X-ray photoelectron spectroscopy (XPS), temperature-programmed reduction (H2-TPR-MS), thermogravimetry (TG) and temperature programmed desorption of ammonia (NH3-TPD) techniques. The catalytic activities were evaluated by the partial oxidation of methanol to dimethoxymethane (DMM). The results showed that vanadia and sulfate were highly dispersed as the catalysts were calcined at 723 and 773 K. The reducibility of the highly dispersed vanadia was stronger than the aggregated vanadia. And the larger number of acidic sites was related to the higher dispersion of sulfate. Moreover, the higher dispersion of vanadia contributed to higher methanol conversion, and the stronger reducibility combined with the larger number of acidic sites led to high DMM selectivity. As a result, the catalysts calcined at 723 and 773 K presented higher methanol conversion and DMM selectivity than those calcined at 673 K or above 823 K.
1. Introduction
Dimethoxymethane (CH3OCH2OCH3, DMM), as an important green downstream product of methanol, is widely used as a diesel additive and building block in organic synthesis. There are numerous reports that DMM can be synthesized by the one-stage oxidation of methanol (3CH3OH + 1/2O2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3OCH2OCH3 + 2H2O).1–10 In these cases, two types of active sites, namely redox sites and acidic sites, are required for DMM synthesis. Here, the redox sites with active lattice oxygen atoms are considered to be involved in the initial formation of formaldehyde (FA) from CH3OH, whereas the acidic sites could catalyze the desired acetalization reactions of FA and CH3OH to DMM. Moreover, the acidic sites were also favorable for dimethyl ether (DME) formation, and the redox sites can catalyze methanol to FA/methyl formate (MF). Therefore, exact matching/cooperation of the redox properties and acidity is critical in getting high DMM selectivity.
CH3OCH2OCH3 + 2H2O).1–10 In these cases, two types of active sites, namely redox sites and acidic sites, are required for DMM synthesis. Here, the redox sites with active lattice oxygen atoms are considered to be involved in the initial formation of formaldehyde (FA) from CH3OH, whereas the acidic sites could catalyze the desired acetalization reactions of FA and CH3OH to DMM. Moreover, the acidic sites were also favorable for dimethyl ether (DME) formation, and the redox sites can catalyze methanol to FA/methyl formate (MF). Therefore, exact matching/cooperation of the redox properties and acidity is critical in getting high DMM selectivity.
Several catalysts, such as heteropoly acids,1,2 SbRe2O6,3 supported rhenium oxide,4 Mo12V3W1.2Cu1.2Sb0.5Ox,9 Cu–ZSM-5,11 FeMo12 and sulfated vanadia–titania,13–16 have been applied in DMM synthesis. Among the reported catalysts, the sulfated vanadia–titania catalysts (VTS) have been considered to be promising for DMM synthesis with high DMM selectivity at lower reaction temperature.13,14 While, the physicochemical characteristics of this catalyst, especially the acidic and redox properties, are strongly related to the existing state of vanadia, sulfate and titania.14,17–19 Generally, the high dispersion of vanadia and sulfate contributes to stronger reducibility and more acidic sites. Moreover, the anatase TiO2 is usually more active than rutile TiO2, due to the fact that the properties of the vanadium species present on the surface of the anatase phase have different acid–basic and redox properties, while those present on the surface of the rutile polymorph phase resemble the properties of those of pure V2O5.20 Therefore, to obtain the best catalytic performance of VTS catalyst in methanol oxidation reaction, it is necessary to optimize the existing state of the vanadia, titania and sulfate. A reasonable and simple approach towards the solution of this problem is related with the optimization of calcination temperature. Although several attempts have been made to improve the catalytic performance of vanadia–titania catalysts by optimizing the calcination temperature,21–23 those for the sulfated vanadia–titania catalysts have received little attention.
In the present study, the VTS catalysts were prepared by the rapid combustion (RC) method and calcined at different temperature. The influence of calcination temperature on the physico-chemical properties of catalysts were studied by XRD, FTIR, BET, ICP-OES, XPS, TG, H2-TPR-MS and NH3-TPD techniques. Moreover, the samples were also tested in the methanol partial oxidation reaction. The aim of this work was to obtain matched redox and acidity to optimize DMM selectivity.
2. Experimental
2.1 Catalyst preparation
The VTS catalysts were prepared by the rapid combustion (RC) method. In the typical synthesis: 13.89 ml TiCl4 was added to 37.86 ml 67 wt% HNO3 under vigorously stirring under N2 atmospheres, and then 6.22 g NH4VO3, 3.37 g Ti (SO4)2 and 73.7 g carbamide were added to the solution, the obtained mixture was stirred until the NH4VO3 and carbamide were completely dissolved, and then calcined under 673, 723, 773, 823 and 873 K, respectively, for 15 min. The obtained catalysts were marked as VTS-673, VTS-723, VTS-773, VTS-823 and VTS-873, respectively. For comparison, the sulfated vanadia–titania sample without calcination and the pure rutile TiO2 calcined at 973 K was prepared by the same method, which were marked as VTS and TiO2-R, respectively.2.2 Catalyst characterization
The specific surface areas of the catalysts were measured by the BET method with a Micromeritics model ASAP 2000 using nitrogen at −196 °C. Prior to measurements, all catalysts were outgassed at 150 °C under 1 × 10−5 Torr residual pressure. XRD patterns were measured on a Bruker Advanced X-ray Solutions/D8-Advance using Cu Kα radiation (λ = 1.5404 Å). The skeletal FT-IR spectra were recorded with a Bruker Vector 22 FTIR spectrophotometer (DTGS detector) operating in the 4000–400 cm−1 range, with a resolution of 2 cm−1 and 100 acquisition scans. In each experiment, 2 mg of sample were mixed with 198 mg of KBr. A spectrum was recorded at room temperature. TG was carried out on a SetaramTGA-92 analyzer with a heating speed of 10 K min−1 in the 303–1073 K range under air in a flow of 50 ml min−1. The elemental analysis (Ti, V, and S) was performed by inductively coupled plasma-optical emission spectroscopy (ICP-OES). The XPS measurements were performed in a Physical Electronics Company Quantum-2000 Scanning ESCA Microprobe equipped with a Al X-ray source (23.3 mA and 1.5 kV), a solid angle acceptance lens and a hemispherical electron analyzer. Samples were compressed in small cup under the pressure of 5 kg cm−2 for 30 s and supported on a holding ceramic carousel. The positive charge, developed on the samples due to the photoejection process was compensated by a charge neutralizer (low energy electron and low energy ion beam). The residual pressure in the spectrometer chamber was 5 × 10−7 Pa during data acquisition. The analyzed area of the sample was 100 μm and the energy region of the photoelectrons were scanned at a pass energy of 29.35 eV. The resolution was 0.68 eV. The binding energies of O 1s, V 2p, Ti 2p and S 2p were referenced to the C 1s band at 284.8 eV. The data was treated on Phi Multipack Program, Gaussian/Lorentzian = 80%. Atomic concentration ratios were calculated by correcting the measured intensity ratios with the manufacturer supplied sensitivity factor. The H2-TPR-MS was carried in continuous mode using a U-type quartz micro-reactor (3.5 mm in diameter). A sample of about 25 mg was contacted with a H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ar mixture (5% H2/Ar) at a total flow rate of 60 cm3 min−1. The inlet and outlet gas compositions were measured using a quadrupole mass spectrometer QMC 311 Balzers. The NH3-TPD spectra were recorded in a fixed-bed reactor system equipped with a gas chromatograph. The catalyst (200 mg) was pretreated at 773 K under Ar flow (60 ml min−1) for 2 h and then cooled to 373 K. Then NH3 was introduced into the flow system. The TPD spectra were recorded at a temperature rising rate of 10 K min−1 from 373 to 900 K.
:Ar mixture (5% H2/Ar) at a total flow rate of 60 cm3 min−1. The inlet and outlet gas compositions were measured using a quadrupole mass spectrometer QMC 311 Balzers. The NH3-TPD spectra were recorded in a fixed-bed reactor system equipped with a gas chromatograph. The catalyst (200 mg) was pretreated at 773 K under Ar flow (60 ml min−1) for 2 h and then cooled to 373 K. Then NH3 was introduced into the flow system. The TPD spectra were recorded at a temperature rising rate of 10 K min−1 from 373 to 900 K.
2.3 Catalyst test
The catalytic oxidation reaction was carried out in a continuous flow fixed-bed reactor containing catalyst (1.0 g) diluted with ground quartz. Before reaction, catalysts were treated in flowing 10% O2/Ar (85 cm3 min−1) for 2 h at 673 K. And then the methanol was introduced into the reaction zone by bubbling O2/Ar (1/9) through a glass saturator filled with methanol (99.9%) maintained at 278 K. The feed composition was maintained as Ar:O2:CH3OH = 84.6:9.4:6.0 (v/v/v). The reaction products were analyzed by on-line gas chromatography (GC-950) using a Propack T column and a TDX-01 column connected to TCD detector and FID detector, respectively. The gas lines were kept at 373 K to prevent condensation of the reactant and products. The reaction was carried out at atmospheric pressure. The product selectivity was calculated on carbon molar base: Si = Yini/∑Yini × 100%, where i is CH3OCH2OCH3, CH3OCH3, HCHO, HCOOCH3, COx, Si is the selectivity of product i, Yi is the number of carbon atom of product i, and ni is the molar of product i.
3. Results and discussion
3.1 Physico-chemical characterization
The textual properties of the catalysts are listed in Table 1. The surface area of the catalysts increased with increasing calcination temperature, and reached the maximum (120.19 m2 g−1) on VTS-773. While, as the calcination temperature continued to increase, the surface area decreased, only 12.58 m2 g−1 on VTS-873.| Catalyst | SBET (m2 g−1) | Binding energy (eV) | Surface compositiona (mol%) | Bulk compositionb (mol%) | No. of SO42− per unit area (nm−2) | Sa/Sb | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O 1s | Ti 2p3/2 | V 2p3/2 | S 2p | V | Ti | S | V | Ti | S | V/Tib | ||||
| a Surface composition calculated from XPS data.b Bulk composition calculated from ICP-OES data. | ||||||||||||||
| VTS-673 | 64.4 | 529.7 (81.23%) | 458.2 | 516.4 | 168.7 | 25.89 | 65.26 | 8.85 | 20.48 | 73.2 | 6.32 | 0.28 | 6.95 | 1.40 |
| 532.1 (18.77%) | ||||||||||||||
| VTS-723 | 112.8 | 529.7 (85.11%) | 458.4 | 517.1 | 168.9 | 27.22 | 66.14 | 6.64 | 20.96 | 73.69 | 5.35 | 0.28 | 3.38 | 1.24 |
| 532.0 (14.89%) | ||||||||||||||
| VTS-773 | 120.2 | 529.8 (87.87%) | 458.3 | 517.1 | 168.9 | 27.68 | 66.54 | 5.78 | 22.31 | 73.1 | 4.59 | 0.31 | 2.74 | 1.26 |
| 532.0 (12.13%) | ||||||||||||||
| VTS-823 | 69.7 | 529.8 (90.36%) | 458.4 | 517.2 | 169.4 | 28.54 | 66.93 | 4.53 | 21.29 | 74.59 | 4.12 | 0.29 | 4.20 | 1.16 |
| 531.9 (9.64%) | ||||||||||||||
| VTS-873 | 12.6 | 529.8 (95.36%) | 458.4 | 517.2 | 169.5 | 30.03 | 67.32 | 2.65 | 22.16 | 75.32 | 2.52 | 0.30 | 14.54 | 1.05 |
| 531.9 (4.64%) | ||||||||||||||
The XRD patterns of the catalysts are shown in Fig. 1. On VTS-673, no diffractions of titania was observed, indicating that the titania might exist as amorphous. While, the typical diffractions of anatase TiO2 (TiO2-A) was observed on VTS-723 and VTS-773. On VTS-823 and VTS-873, the TiO2-A and rutile TiO2 (TiO2-R) were coexisted. While, the intensities of TiO2-R diffraction peaks were stronger on VTS-873 than VTS-823.24 This result indicated that the increasing temperature caused the transformation of TiO2-A to TiO2-R. Moreover, no diffraction peaks of crystalline V2O5 were observed as the calcination temperature was below 823 K, implying that the vanadia was highly dispersed or the V2O5 crystalline was less than 4 nm.14 The typical diffraction peaks of V2O5 crystalline14,25,26 appeared on VTS-873, which indicated the aggregation of vanadia.
 | ||
| Fig. 1 The XRD patterns of the catalysts. | ||
To further study the existing state of vanadia, the FTIR spectra was carried out (see Fig. 2). For VTS-673, VTS-723 and VTS-773 a threshold around 1000 cm−1 were detected, which could be assigned to the bands for polyvanadate species (V–O–V) bonded to the surface of TiO2.14 While, for VTS-823 and VTS-873 a band at 1022 cm−1 due to the microcrystalline V2O5 appeared, simultaneously with the decrease of the peak intensity at 1000 cm−1, suggesting the formation of crystalline V2O5.14–17,25,26 Combined with the XRD results, it can be deduced that the vanadia should be highly dispersed on VTS-673, VTS-723 and VTS-773, while aggregated as crystalline V2O5 on VTS-823 and VTS-873. In addition, a typical band at 1137 cm−1 corresponding to SO42− (ref. 27) was detected for all of the samples, which indicated that the S existed as sulfate. While, its intensity weakened with the increase of calcination temperature, implying the decrease of sulfate content.
 | ||
| Fig. 2 The FT-IIR spectra of the catalysts. | ||
Table 1 lists the binding energies (BE) as well as the surface and bulk composition from XPS and ICP-OES. The binding energies of Ti 3d5/2 for the catalysts were 458.2–458.6 eV, which suggested the presence of Ti4+.27–29 The binding energies of V 2p3/2 for the catalysts were 517.1, 516.4, 517.2 eV, indicated the surface vanadia species were fully oxidized (oxidation state V5+).29 The common peaks of O 1s for both the catalysts were 529.6 and 532 eV, which belonged to the oxide oxygen and sulfate oxygen, respectively.27 The binding energies at 168.7–169.5 eV (see Fig. 3.) was measured for S 2p3/2, which are typical of sulfur in the S6+ oxidation state, as in Na2SO4 or Fe2(SO4)3.30
 | ||
| Fig. 3 XPS spectra of S 2p for the catalysts. | ||
The analysis of bulk and surface sulfate content showed that both the surface and bulk sulfate content decreased with the increase of calcination temperature, which might be due to the decomposition of sulfate (see TG result). Moreover, the surface sulfate content was higher than that of bulk, indicating that the sulfate mainly existed on the surface of the catalysts. Based on the surface area and the sulfate content, the number of S (atoms per nm2) was calculated. The values for VTS-723 and VTS-773 were 3.38 and 2.74, respectively. While, those were 6.95, 4.20 and 14.54 for VTS-673, VTS-823 and VTS-873, respectively. According to the reported result,32 the necessary mole concentration of sulfate to form a monolayer was 3.9 S per nm2. Thus, the sulfate was highly dispersed on VTS-723 and VTS-773, while aggregated on VTS-673, VTS-823 and VTS-873.
The H2-TPR-MS was carried out to confirm the existing state of sulfate (in Fig. 4). Three types of product, H2O, SO2 and H2S, were detected with their formation temperatures in parallel with the hydrogen consumption. According to the literature, the formation of H2O is due to the reduction of VOx, while the SO2 and H2S are due to the reduction of sulfate.32 For VTS-673, VTS-823 and VTS-873, only SO2 was detected (see Fig. 4a, d and e). While, for VTS-723 and VTS-773 (see Fig. 4b and c), both H2S and SO2 were detected. It was reported31,32 that the production of SO2 and H2S during H2 reduction was correspond to the reduction of bidentate sulfate species and the emission of only SO2 was due to the reduction of pyrosulfate species. Combined with the present study, it was clear that the sulfate dispersed as bidentate sulfate on VTS-723 and VTS-773, while aggregated as pyrosulfate sulfate on other samples. In addition, the intensity of SO2 on VTS-673 was stronger than those on VTS-823 and VTS-873, which might be due to the decomposition of sulfate on VTS-823 and VTS-873 (see TG result).
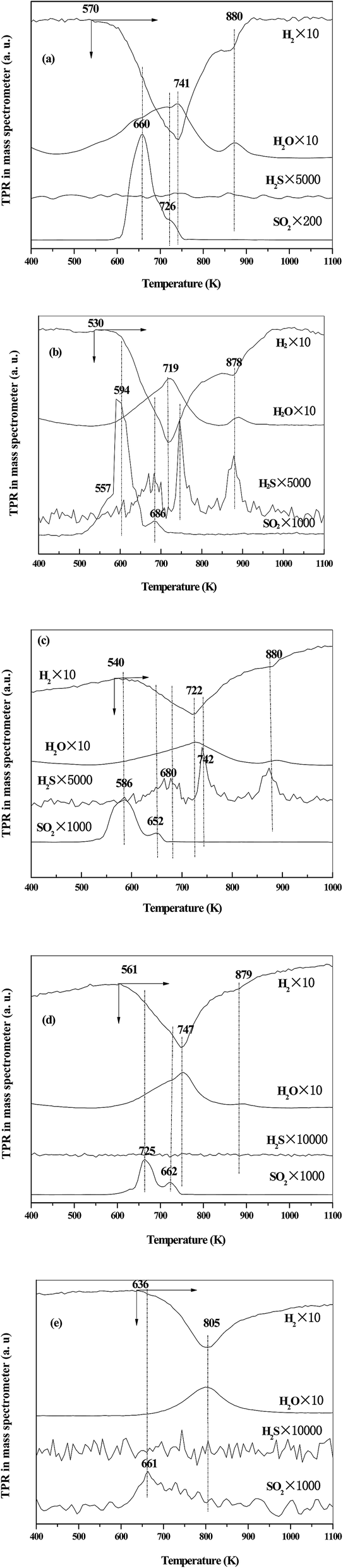 | ||
| Fig. 4 The H2-TPR-MS profiles of the catalysts: (a) VTS-673, (b) VTS-723, (c) VTS-773, (d) VTS-823 (e) VTS-873. | ||
To further study the reasons leading to the decreasing content of sulfate, the thermal stability of sulfate was characterized by TG on VTS (see Fig. 5). The VTS sample exhibited four weight losing peaks at 379, 635, 768 and 884 K, respectively. The peak at 379 K is attributed to the removal of water, while those at 635, 768 and 884 K are corresponding to the decomposition of sulfate.31 Combined with the above results, it can be deduced that the decreased content of sulfate was due to the decomposition of sulfate under high temperature.
 | ||
| Fig. 5 The TG profiles of the VTS catalyst. | ||
3.2 Redox and acidity properties
In H2-TPR process, the profiles of hydrogen consumption could reflect the redox property of metal oxide.31 As shown in Fig. 4, for VTS-673, a major reducible peak was observed at 741 K, which shifted to 720 K on VTS-723 and VTS-773, while shifted to 805 K on VTS-873. This result indicated that the catalysts calcined at 723 and 773 K exhibited stronger reducibility than those calcined at 673 K or above 823 K. The different reducibility of the catalysts could be due to the different existing state of vanadia. For VTS-723 and VTS-773, the stronger reducibility was due to the high dispersion of vanadia. While, for VTS-823 and VTS-873, the appearance of crystalline V2O5 led to poor reducibility.The acidity of the catalysts was characterized by NH3-TPD technique (see Fig. 6 and Table 2). All of the catalysts showed similar feature of NH3 desorption, which spread in the temperature range of 400–700 K, indicating the acidity strength of the samples was similar. However, the number of acidic sites among the catalysts was significantly different. For VTS-723 and VTS-773, the values were 745 and 817 μmol g−1, which were much larger than those on VTS-673, VTS-823 and VTS-873. To quantitatively analyze the effect of the type of sulfate species on the number of acidic sites, the number of absorbed ammonia per sulfate species was calculated. The values were 1.18 and 1.50 for VTS-723 and VTS-773, and only 0.78, 0.79 and 0.89 for VTS-673, VTS-823 and VTS-873. As a result, the high dispersion of sulfate on VTS-723 and VTS-773 led to larger number of acidic sites, and the aggregation of sulfate on VTS-673, VTS-823 and VTS-873 resulted in small number of acidic sites.
 | ||
| Fig. 6 The NH3-TPD profiles of the catalysts. | ||
| Catalyst | No. of acidic sites (μmol g−1) | No. of acidic sites per sulfate species |
|---|---|---|
| VTS-673 | 579.99 | 0.78 |
| VTS-723 | 745.49 | 1.18 |
| VTS-773 | 817.94 | 1.50 |
| VTS-823 | 384.80 | 0.79 |
| VTS-873 | 266.90 | 0.89 |
3.3 Catalytic performance
Fig. 7 shows the influence of calcination temperature on methanol conversion. As it can be seen, the VTS-723 and VTS-773 were more active than VTS-673, VTS-823 and VTS-873. For example, at 403 K, the methanol conversion was only 19.51% on VTS-673, while increased up to 43.53% and 42.72% on VTS-723 and VTS-773, and then decreased to 0% on VTS-873. The catalytic performances at other temperatures were similar to 403 K. The previous reports14,17 showed that the catalytic activity of vanadia-based catalyst in methanol oxidation reaction is closely related to the existing state of vanadia. And the highly dispersed vanadia contributes to high activity, while the V2O5 crystalline exhibits low activity.33–36 Combined with the XRD and FTIR results, it can be deduced that the high dispersion of vanadia on VTS-723 and VTS-773 contributed to high methanol conversion. On VTS-823 and VTS-873, the appearance of crystalline V2O5 and the appearance of TiO2-R led to the low methanol conversion. | ||
| Fig. 7 Effect of calcination temperature on methanol conversion. | ||
Table 3 shows the effect of calcination temperature on DMM selectivity at similar methanol conversion (about 45%). The DMM selectivity showed similar changing trend to methanol conversion, and reached the maximum on VTS-723 and VTS-773. That phenomenon could be explained by the difference of acidity and redox properties among these catalysts. According to the mechanism of the one-step synthesis of DMM from methanol reported by Liu,1 the methanol was primarily oxidized to FA on redox sites, and then the methanol reacted with FA to form DMM on acidic sites. Furthermore, our previous study33,34 showed that the higher DMM selectivity on the vanadium-based catalysts was due to the stronger reducibility matched with larger number of the acidic sites. Combined with the present study, it can be deduced that the stronger reducibility of VTS-723 and VTS-773 could enhance the oxidation of methanol to FA, while the more number of acidic sites could enhance the acetalization reactions of FA and CH3OH to DMM and finally lead to high DMM selectivity. For catalysts calcined at 673 K or above 823 K, the poor reducibility and small number of acidic sites could not efficiently catalyze the first and the second reaction, leading to the low DMM.
| Catalyst | Selectivity (%) | ||||
|---|---|---|---|---|---|
| FA | DME | MF | DMM | COx | |
| a Reaction condition: Ar/O2/CH3OH = 84.6/9.4/6.0 (v/v/v), 393 K, at about 45% methanol conversions. | |||||
| VTS-673 | 20.35 | 3.36 | 8.92 | 66.64 | 0.73 |
| VTS-723 | 3.23 | 0.94 | 5.16 | 90.33 | 0.34 |
| VTS-773 | 1.98 | 0.57 | 5.63 | 91.76 | 0.07 |
| VTS-823 | 7.26 | 1.10 | 8.28 | 83.21 | 0.15 |
| VTS-873 | 19.90 | 1.23 | 14.09 | 64.46 | 19.90 |
Conclusion
The calcination temperature has profound influence on catalyst structure, which in turn affected the acidity and reducibility as well as catalytic performance in the one-step oxidation of methanol to DMM. The highest dispersion of vanadia and sulfate was obtained on VTS-723 and VTS-773. The stronger reducibility and more acidic sites were closely related to highly dispersed vanadia and sulfate, while the poor reducibility and small acidic sites were relate to the aggregated vanadia and sulfate. The highest activity and DMM selectivity obtained on VTS-723 and VTS-773 was due to the highest dispersion of vanadia and the corresponding stronger reducibility and large number of acidic sites.Acknowledgements
The authors gratefully acknowledge the Key Project of Natural Science Foundation of China (No. 21303241) and Key Project of Natural Science Foundation of Shanxi Province (No. 2012021005-6).Notes and references
- H. C. Liu and E. S. Iglesia, J. Phys. Chem. B, 2003, 107, 10840 CrossRef CAS.
- H. C. Liu and E. S. Iglesia, J. Catal., 2004, 223, 161 CrossRef CAS PubMed.
- Y. Z. Yuan, H. C. Liu, H. Imoto, T. Shido and Y. Iwasawa, J. Catal., 2000, 195, 51 CrossRef CAS.
- Y. Z. Yuan, T. Shido and Y. Iwasawa, J. Phys. Chem. B, 2002, 106, 4441 CrossRef CAS.
- H. C. Liu and E. S. Iglesia, J. Phys. Chem. B, 2005, 109, 2155 CrossRef CAS PubMed.
- G. A. Busca, S. Elmi and P. J. Forzatti, J. Phys. Chem., 1987, 91, 5236 CrossRef.
- C. R. Deltcheff, A. Aouissi, S. Launayb and M. Fournier, J. Mol. Catal. A: Chem., 1996, 33, 114 Search PubMed.
- I. J. Shannon, T. Maschmeyer, R. D. Oldroyd, G. Sankar and J. M. Thomas, J. Chem. Soc., Faraday Trans., 1998, 94, 1495 RSC.
- S. Royer, X. Sécordel, M. Brandhorst, F. Dumeignil, S. Cristol, C. Dujardin, M. Capron, E. Payena and J. L. Dubois, Chem. Commun., 2008, 86, 57 Search PubMed.
- H. Q. Guo, D. B. Li, H. C. Xiao, J. L. Zhang, W. H. Li and Y. H. Sun, Korean J. Chem. Eng., 2009, 26, 902 CrossRef CAS.
- Y. H. Zhang, I. J. Drake, D. N. Briggs and A. T. Bell, Synthesis of dimethyl carbonate and dimethoxy methane over Cu-ZSM-5, J. Catal., 2006, 244, 219 CrossRef CAS PubMed.
- J. L. Gornay, X. Secordel, G. Tesquet, B. D. Menorval, S. Cristol, P. Fongarland, M. Capron, L. Duhamel, E. Payen, J. L. Dubois and F. Dumeignil, Green Chem., 2010, 12, 1722 RSC.
- Y. C. Fu and J. S. Shen, Chem. Commun., 2007, 72, 2121 Search PubMed.
- Q. Sun, Y. C. Fu, J. W. Liu, A. Auroux and J. Y. Shen, Appl. Catal., A, 2008, 334, 26 CrossRef CAS PubMed.
- J. W. Liu, Y. C. Fu, Q. Sun and J. Y. Shen, Microporous Mesoporous Mater., 2008, 116, 614 CrossRef CAS PubMed.
- S. Chen, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Commun., 2011, 47, 9345 RSC.
- F. Roozeboom, T. Fransen, P. Mars and P. J. Gellings, Z. Anorg. Allg. Chem., 1979, 449, 25 CrossRef CAS PubMed.
- G. Hausinger, H. Schmelz and H. Knijzinger, Appl. Catal., 1988, 39, 267 CrossRef CAS.
- M. P. Casaletto, L. Lisi, G. Mattogno, P. Patrono, F. Pinzari and G. Ruoppolo, Catal. Today, 2004, 91, 271 CrossRef PubMed.
- B. G. Swierkoz, Appl. Catal., A, 1997, 157, 263 CrossRef.
- R. Y. Saleh, I. E. Wachs, S. S. Chan and C. C. Chersich, J. Catal., 1986, 98, 102 CrossRef CAS.
- M. A. Bañares, L. J. Alemanya, M. C. Jiménez, M. A. Larrubi, F. Delgado, M. L. Granados, A. Ma. Nez-Arias, J. M. Blasco and J. L. G. Fierro, J. Solid State Chem., 1996, 124, 69 CrossRef.
- P. Oliveira, M. L. Rojas-Cervantes, A. M. Ramos, I. M. Fonseca, A. M. Botelho do Rego and J. Vital, Catal. Today, 2006, 118, 307 CrossRef CAS PubMed.
- M. A. Banares, L. J. Alemany, M. C. Jiménez, M. A. Larrubia, F. Delgado, M. L. Granados, A. Martĺnez-Arias, J. M. Blasco and J. L. G. Fierro, J. Solid State Chem., 1996, 124, 69 CrossRef CAS.
- H. Y. Zhao, S. Bennici, J. X. Cai, J. Y. Shen and A. Auroux, J. Catal., 2010, 274, 259 CrossRef CAS PubMed.
- H. Y. Zhao, S. Bennici, J. X. Cai, J. Y. Shen and A. Auroux, Appl. Catal., A, 2010, 385, 224 CrossRef CAS PubMed.
- J. P. Chen and R. T. Yang, J. Catal., 1993, 139, 277 CrossRef CAS.
- J. P. Nogier and M. Delamar, Catal. Today, 1994, 20, 109 CrossRef CAS.
- V. I. Bukhtiyarov, Catal. Today, 2000, 56, 403 CrossRef CAS.
- M. Bensitel, O. Saur and J. C. Lavalley, Mater. Chem. Phys., 1988, 19, 147 CrossRef CAS.
- L. Baraket, A. Ghorbel and P. Grange, Appl. Catal., B, 2007, 72, 37 CrossRef CAS PubMed.
- J. B. Laizet, A. K. Søiland, J. Leglise and J. C. Duchet, Top. Catal., 2000, 10, 89 CrossRef CAS.
- H. Q. Guo, D. B. Li, D. Jiang, W. H. Li and Y. H. Sun, Catal. Today, 2010, 158, 439 CrossRef CAS PubMed.
- Z. H. Fan, H. Q. Guo, K. G. Fang and Y. H. Sun, RSC Adv., 2015, 5, 24795 RSC.
- K. V. R. Chary, G. Kishan, C. P. Kumar, U. V. Sagar and J. W. Niemantsverdriet, Appl. Catal., A, 2003, 245, 303 CrossRef CAS.
- A. S. Elmi, E. Tronconi, C. Cristiani, J. P. Gomez Martin and P. Forzatti, Ind. Eng. Chem. Res., 1989, 28, 387 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |