DOI:
10.1039/C5RA08778C
(Paper)
RSC Adv., 2015,
5, 65496-65513
Antihypertensive activity of a quinoline appended chalcone derivative and its site specific binding interaction with a relevant target carrier protein†
Received
11th May 2015
, Accepted 24th July 2015
First published on 24th July 2015
Abstract
Inhibition of Angiotensin Converting Enzyme (ACE) is identified as a main therapeutic target in controlling hypertension. The principal intent of this work is to investigate the ACE inhibitory property of a quinoline appended chalcone derivative (E)-3-(anthracen-10-yl)-1-(6,8-dibromo-2-methylquinolin-3-yl)prop-2-en-1-one (ADMQ), and its binding mechanism with model transport protein BSA by employing steady state and time resolved fluorescence, Circular Dichroism (CD), in silico molecular docking, Induced Fit Docking (IFD) and Molecular Dynamics (MD) simulation. Incubation of ADMQ with kidney cortex plasma membrane shows considerable antihypertensive effect by the inhibition of ACE. ADMQ undergoes strong interaction with ACE both in the absence and presence of BSA. Comparable ACE inhibitory mechanistic profile of ADMQ with standard drug captopril has been identified in terms of the ligand interaction pattern, changes in secondary structural elements and protein RMSF. The steady state emission of BSA undergoes a remarkable decrement via ground state complex formation upon addition of ADMQ in an aqueous buffer solution of BSA at a physiological pH of 7.4 contrary to the time resolved and FRET measurement where both the static and energy transfer mechanism co-exists. The rotationally restricted ADMQ molecule shows strong binding affinity towards subdomain IIA of site I with a close proximity (2.45 nm) to the Trp 213 residue. The minor decrease of α-helical content as calculated from CD spectral measurement and 1–3 Å change in protein RMSD during MD simulation clearly indicate that the polypeptide chain is partially destabilized due to the above site specific accommodation of the host (ADMQ). A slight diminution in the ACE inhibitory profile is observed in the presence of BSA; however BSA shows lesser binding towards ADMQ in the presence of the target enzyme. The spectroscopic research described herein may provide enormously important information for ACE inhibition of the chalcone derivative and its detailed binding interaction with a carrier protein for chalcone based drug designing in medicinal chemistry research.
Introduction
There has been an increasing interest in chalcone based drug designing due to growing evidence of their versatile health benefits.1–4 Chalcones (α,β-unsaturated ketones) are open chain flavonoids and are biogenetic precursors of flavonoids in higher plants. They are intermediates and end products of flavonoid biosynthesis and starting material for the synthesis of different types of highly bioreactive heterocyclic compounds, such as pyrazolines, pyrimidines, quercetins etc.5 Consumption of plants and plant products that are rich in flavonoids has been related with protective effects against cardiovascular disease.6,7 Amongst the foremost threat to cardiovascular and renal disorders, hypertension is a widespread and frequently progressive ailment. Enormous efforts have occurred for the synthesis of innovative antihypertensive agents to combat this lethal disease. Among the many classes of antihypertensives, angiotensin converting enzyme (ACE) inhibitors are commonly used to treat cardiovascular diseases such as high blood pressure, heart failure, coronary/artery disease and kidney failure which inhibits the conversion of angiotensin I to angiotensin II. Chalcones are class of antihypertensive drugs that seeks to prevent the complications of high blood pressure.8,9 Heterocyclic chalcone scaffolds in this regard have attracted considerable attention as they are widely used in medicinal chemistry research and have shown antihypertensive activity through inhibition of ACE.10 The amino alkoxychalcones and their pharmaceutically acceptable acid addition salts possess superior pharmacological antihypertensive and vasodilative activities.11
Serum Albumins (SAs) are known to have a number of specific binding sites and have a major functional role as versatile transporters of various drugs through the blood stream via their initial temporary boarding into pocket. This transportation mechanism depends on the conformational ductility and adjustability of the protein (host) to facilitate the interaction with the drug (guest).12–14 BSA and Human Serum Albumin (HSA) are the most studied serum proteins in pharmaceutical industry due to their ability to bind various drug molecules and alter their pharmacokinetic/pharmacodynamic properties.15,16
BSA constitutes three homologous domains (I, II, III) which are divided into nine loops (L1–L9) by 17 disulfide bonds.17 X-ray crystallographic data show that the albumin structure is predominantly α-helical with the remaining polypeptide occurring in turns and extended or flexible regions between subdomains with no β-sheets. It has two tryptophan residues that possess intrinsic fluorescence: Trp 134 in the first and Trp 213 in the second domain, located on the surface of the molecule and hydrophobic binding pocket of the protein respectively.18 The specific delivery of a drug originates from the presence of two major and structurally selective binding sites, namely Sudlow site I and site II, which are located in three homologous domains that form a heart-shaped protein.19 In BSA, the binding affinity offered by site I is mainly through hydrophobic interactions, whereas site II involves a combination of hydrophobic, hydrogen bonding, and electrostatic interactions.20,21 The strength of binding of a specific ligand/drug molecule at either of these sites is very much dependent on the nature of amino acid residues available there.22 Also, the pocket specific interactions of BSA is different for different exogenous ligands. The reception offered by the host will also differ due to the nature of the ligand and the capability of the protein to accommodate the ligand.23 Whenever a ligand is accommodated in the protein, the response could be the structural perturbation in that region of macromolecule tendered by the guest. The conformational changes of serum albumin that is induced by its interaction with low molecular-weight drugs may affect the biological function of serum albumins as the carrier protein.24
In this present article the antihypertensive activity of a quinoline appended chalcone derivative (in presence and absence of model transport protein) and its detail interaction with the target enzyme as well as with carrier protein have been ascertained using UV-visible absorption, steady state and time resolved fluorescence spectroscopy, fluorescence anisotropy, and circular dichroism (CD) techniques. The experimental observations have further been rationalized with in silico molecular docking and molecular dynamics simulation. The consolidated ‘in vitro’ and ‘in silico’ spectroscopic research described herein may provide significant information concerning the usefulness of a heterocyclic chalcone derivative as an effective antihypertensive agent along with its mechanistic details of ACE inhibition in terms of ligand interaction pattern, SSE and protein RMSF, additionally it may provide substantial information regarding its transportation mechanism. These information could be useful for rational chalcone based drug design in terms of pharmacokinetics and pharmacodynamics.
Results and discussions
Antihypertensive effect of ADMQ
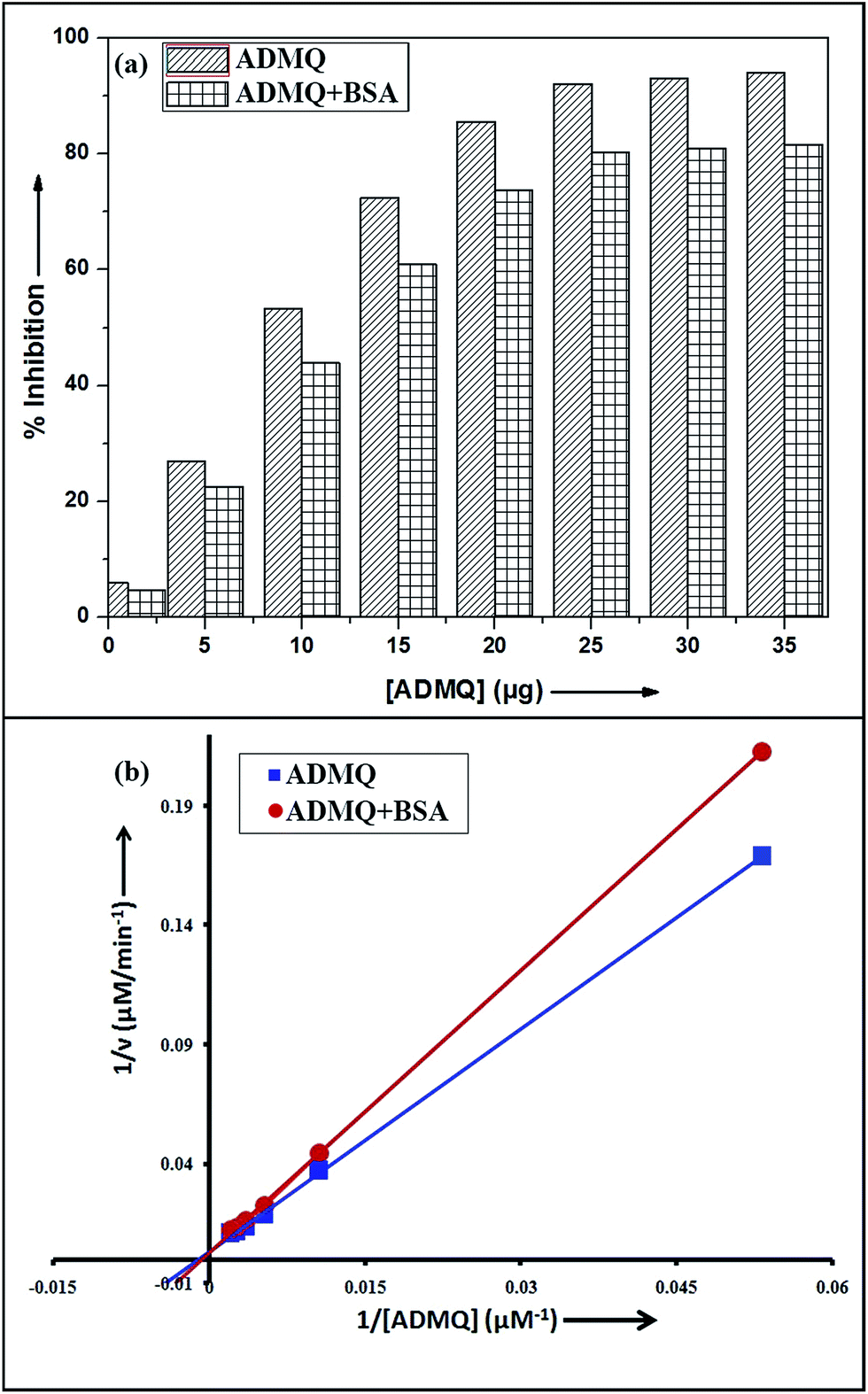
Among the many classes of therapeutic antihypertensive agents, ACE inhibitors are most commonly used to treat cardiovascular diseases such as hypertension. ACE, a zinc dependent peptidase catalyzes the conversion of angiotensin I to angiotensin II (Ang II)25 and the overproduction of Ang II leads to hypertension. So the inhibition of ACE has been used as one of the methods for the treatment of hypertension related to cardiovascular disorders and congestive heart failure.26 To verify its antihypertensive activity, ADMQ is incubated with the kidney cortex plasma membrane (enzyme) for 10 min at 37 °C followed by the incubation with Hippuryl-His-Leu (substrate) for 45 min at 37 °C which developed a yellow colour by addition of pyridine and benzene sulphonyl chloride. The colour formed has been measured at 410 nm in ELISA plate reader. The probe ADMQ has been screened at eight different concentrations 1, 5, 10, 15, 20, 25, 30 and 35 μg/100 μl that blocks the substrate availability to the enzyme and causes ACE inhibition in order of 92% at tested concentrations as shown in Fig. 1a. Inhibitory constant27,28 obtained is found to be of the order of 167.19 μM (Fig. 1b).
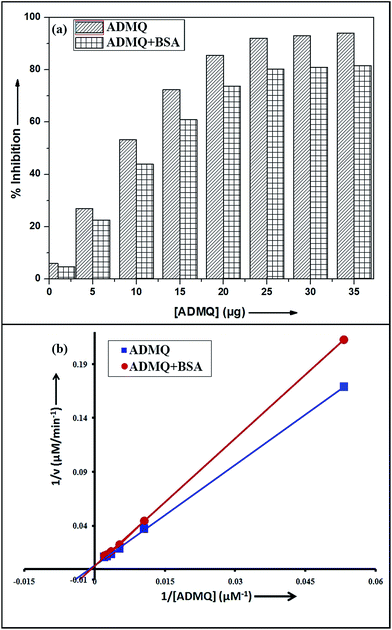 |
| | Fig. 1 (a) ACE inhibitory profile of ADMQ and ADMQ + BSA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). (b) Lineweaver–Burk plot for inhibition of ACE in absence and presence of BSA. :1). (b) Lineweaver–Burk plot for inhibition of ACE in absence and presence of BSA. | |
Identification of binding interaction of ADMQ with ACE
Steady state absorption and emission spectroscopy. Interaction of ADMQ with angiotensin converting enzyme has been studied using electronic absorption, steady state emission and time resolved fluorescence spectroscopic techniques. Since ACE does show considerable emission at tested concentration, the above said interaction has been monitored by probing ADMQ molecule. In aqueous buffer solution at pH 7.4, the probe ADMQ shows two distinct absorption bands at 259 nm and 434 nm. The more intense peak at 259 nm arises from mixed contribution from several configurations such as HOMO−1 to LUMO whereas the red shifted band with lower oscillator strength arises due to HOMO to LUMO transition.29 On gradual addition of ACE in aqueous buffer solution of probe molecule, both the absorption band decreases significantly and more interestingly the lower energy absorption band is slightly (4–5 nm) blue shifted (Fig. 2a). This may be an indication of localization of the chromophore ADMQ in a different microenvironment (as compared to the bulk aqueous solution) along with a specific ground state interaction with target enzyme.
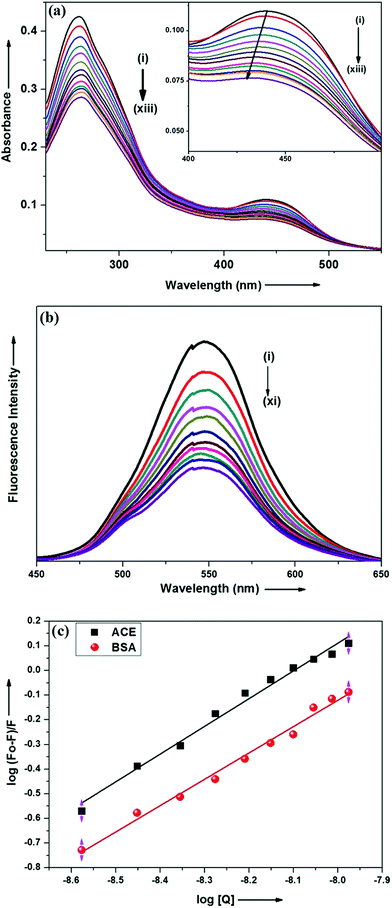 |
| | Fig. 2 (a) Absorption spectra of ADMQ with gradual addition of ACE at pH 7.4 at 298 K. Curves (i)–(xiii) correspond to [ACE] = 0.0, 0.88, 1.76, 2.65, 3.53, 4.41, 5.29, 6.17, 7.05, 7.93, 8.81, 9.69 and 10.5 × 10−9 mol dm−3 respectively. [ADMQ] = 9.87 × 10−6 mol dm−3. (b) Fluorescence spectra of ADMQ on gradual addition of ACE at 298 K at 0.05 M HEPES buffer at pH 7.4. λexc = 434 nm. Curves (i)–(xi) correspond to [ACE] = 0.0, 0.88, 1.76, 3.53, 4.41, 5.29, 6.17, 7.05, 7.93, 8.81 and 10.5 × 10−9 mol dm−3 respectively. (c) Effect of ACE and BSA on ADMQ in 0.05 M HEPES buffer of pH 7.4 at 298 K λexc = 434 nm, λem = 550 nm. | |
The above said ground state interaction has further got its confirmation from the emission measurement. ADMQ exhibits considerable emission at 550 nm while exciting at its lowest energy band. On gradual addition of ACE in the aqueous buffer solution of ADMQ, the emission intensity decreases without any significant spectral shift of the emission maxima (Fig. 2b). This diminution of the fluorescence intensity may be attributed to the binding of ADMQ with ACE. The binding interaction of ADMQ–ACE system has been quantified by the following eqn (1)22,30,31
| | |
logF0 − F/F = logKb + nlog[Q]
| (1) |
where
F0 is the fluorescence intensity of ADMQ alone,
F is the fluorescence intensity in presence of ACE and
Q is the concentration of ACE. The binding constant (
Kb) for ADMQ–ACE system is found to be 1.27 × 10
9 (±10%) at 298 K and pH 7.4, (
Fig. 2c). This calculated binding constant clearly indicates a strong binding interaction between probe and the enzyme molecule.
Time resolved fluorescence measurement
To substantiate the above said binding interaction, the time resolved fluorescence decay profile of ADMQ has been monitored with increase in the enzyme concentration. In aqueous buffer solution (pH = 7.4) ADMQ shows biexponential fluorescence decay with life time 955 ps (τ1) and 3.89 ns (τ2) respectively. On gradual addition of ACE, both the fluorescence lifetime (τ1) and (τ2) decrease slightly (Fig. S1, Table S2†). This observation points toward the static quenching mechanism with no perturbation of the exited states, while the emission is exclusively coming from the “free” ADMQ molecule (that have not formed the complex with ACE), and as soon as the number of ACE molecule increases, there are lesser number of the free ADMQ molecule exist to emit as it is exactly imitated from the profound diminution in the steady state emission measurement. However the tiny decrement in the lifetime value may be because of the slight change in the microenvironment as indicated in the spectral shift in the absorption spectra. Hence time resolved fluorescence measurement supports the UV-visible absorption and steady state emission finding and confirms the ground state complex formation of AQMQ with ACE.
Molecular docking
The ‘in silico’ molecular docking and molecular dynamics simulation have been carried out using X-ray crystal structure of ACE (PDB ID: 1UZF)26 to identify the molecular mechanism of ACE inhibition by ADMQ in comparison with therapeutically used standard antihypertensive drug, captopril. Captopril contains two centers of dissymmetry, one associated with the (S)-proline portion and the other associated with the 3-mercapto-2-methyl-propionic acid side chain.16 The crystal structure of ACE–captopril complex reveals that proline residue of captopril interacts with the S2′ subsite of enzyme (ACE) active site. This interaction is favored by two histidine residues at the enzyme active site: His 513 and His 353. It is also shown that the thiol group of captopril interacts with zinc(II) ion to form zinc thiolate complex.32 Other amino acids that make up the active site are Glu 162, Gln 281, Asp 377, Lys 511, Phe 512 and Tyr 520.
It is relevant to mention here that ADMQ undergoes a structural change (Scheme 1) from its native form (Form I) to β-hydroxyketo form (Form III) in physiological condition.29,33
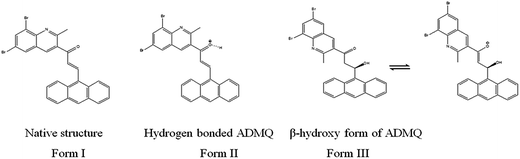 |
| | Scheme 1 Structural change of ADMQ in different environments.29 | |
Hence the molecular docking studies has been carried out with β-hydroxy form of ADMQ to assess its preferred orientation and detailed mechanism of the interaction in the chosen binding site of ACE using Glide programme [Glide, version 6.3, Schrödinger, LLC, NY, 2014] which is used in structure based drug designing, to predict the binding affinity and to optimize small molecules as drug candidates, where flexible ligand is docked to a rigid receptor binding pocket.34 The probe ADMQ is docked in the same active site of ACE which is preferred by captopril, to identify the molecular interaction of the probe with the binding site of the enzyme. The binding affinity of captopril and ADMQ has been compared on the basis of GLIDE score. The GLIDE protocol yielded a score of −6.104 kcal mol−1 for β-hydroxyketo form of ADMQ and −5.401 kcal mol−1 for the standard drug captopril. The comparable Glide score indicate considerable stability for both the compounds in the active site of ACE.
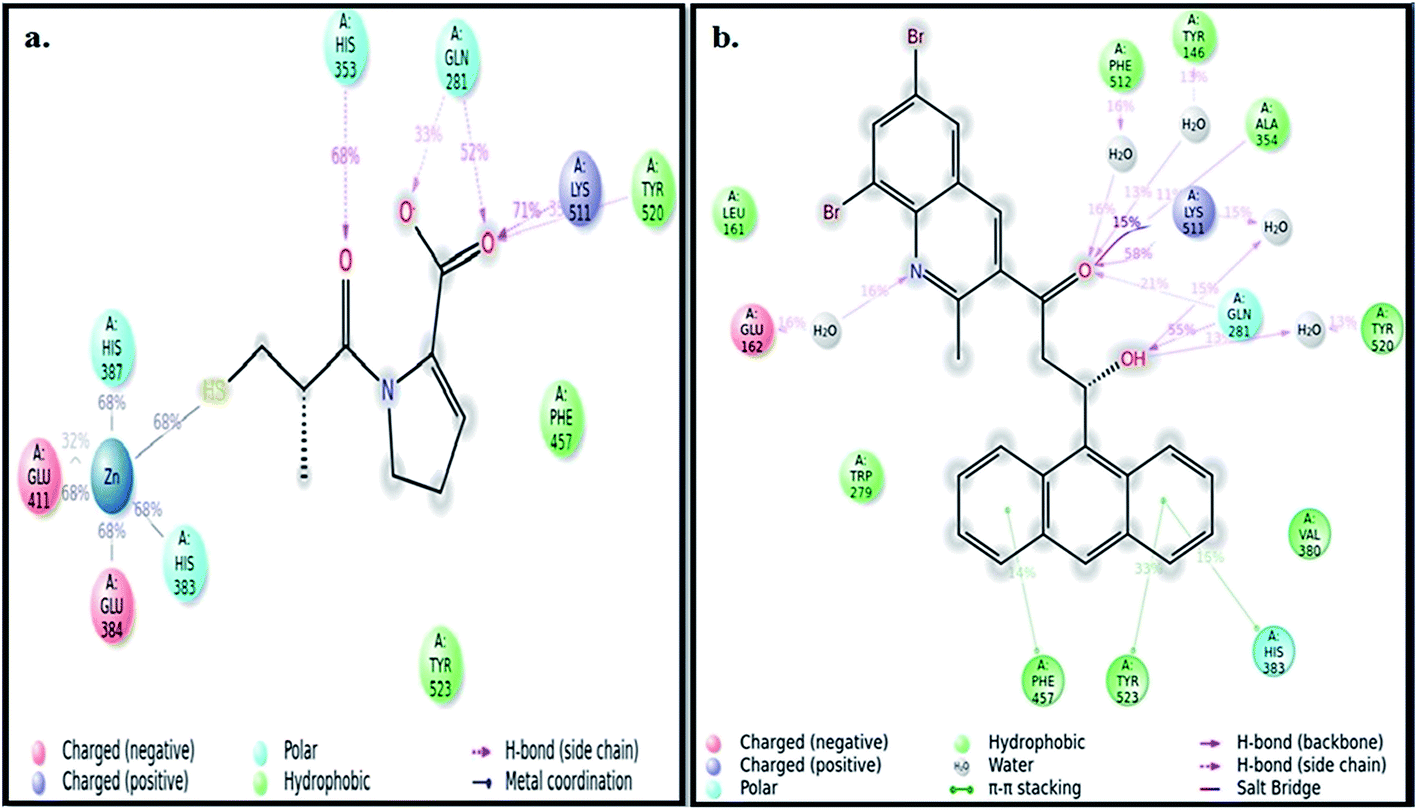
The best pose of both ADMQ and captopril obtained from molecular docking study with ACE have been considered further for the explicit molecular dynamics (MD) simulation by Desmond, where system builder used explicit aqueous medium followed by complex minimization to bring down complex to the least energy level.35 The captopril–ACE and ADMQ–ACE complex real time simulation of 10 ns elucidates the stability and the interaction pattern of the respective probe molecule in terms of the percentage of contacts, hydrogen bonds, hydrophobic, ionic and water bridges (Fig. 3a and b respectively). ADMQ is surrounded by hydrophobic residues inside the pocket of ACE. β-Hydroxy group of ADMQ forms H-bonded with Gln 281 and a bridged water H-bond is formed with Tyr 520. The carbonyl oxygen of the probe is H-bonded with Lys 511 and Ala 354 along with a bridged water H-bond with Phe 512 and Tyr 146. On the other hand (S)-proline portion of captopril forms a H-bond with Gln 281, Tyr 520 and Lys 511, and the carbonyl oxygen of 3-meracapto-2-methyl propionic acid side chain forms hydrogen bond with His 353. From Fig. 3a and b, it is clear that when ADMQ and captopril are docked in the same active site of ACE, both share almost similar interactions via H-bonding (Lys 511, Gln 281 and Tyr 520) and polar group residue like His 383. ADMQ forms π–π stacking interactions with hydrophobic residues (Phe 457 and Tyr 523) whereas captopril is also surrounded by the same hydrophobic amino acid residues. Moreover, in ligand–receptor binding, the cumulative effect of overall interactions (hydrophobic, electrostatic, van der Waals energy and H-bonding) is considered for the energy minimized pose. This cumulative effect is characterized in terms of comparable Glide score34 (binding affinity of different ligands) as mentioned above.
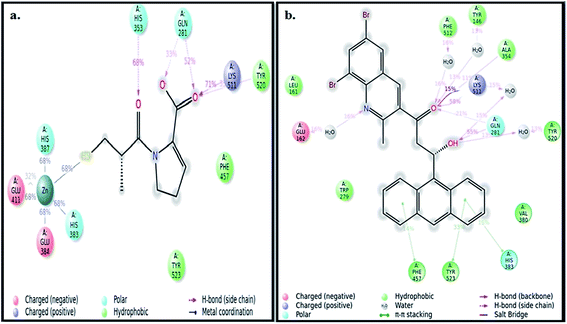 |
| | Fig. 3 (a) Ligand interaction pattern with percentage of contacts with the ligand captopril during 10 ns explicit MD simulation. (b) Ligand interaction pattern with percentage of contacts with the ligand β-hydroxyketo form of ADMQ with ACE during 10 ns explicit MD simulation. | |
In order to investigate the conformational changes in the active site of ACE due to probe binding, the secondary structure elements (SSE) like alpha helices, beta strands, turns and loops have been monitored throughout the simulation trajectory. It has been noticed that the secondary structural elements of ACE in case of captopril (Fig. 4a, top left) as well as ADMQ (Fig. 4a, top right) shows almost similar changes in α-helix and β-strands which indicate that the active site of ACE underwent almost similar conformational changes when both of them are incorporated in the same active site. The local changes along the protein chain have also been identified by the protein Root Mean Square Fluctuation (RMSF) where the peak(s) indicates the areas of protein (residue index) that fluctuate maximum during simulation. For both captopril (red) and ADMQ (blue) (Fig. 4b), the involvement of similar amino acid residues of ACE have been identified.
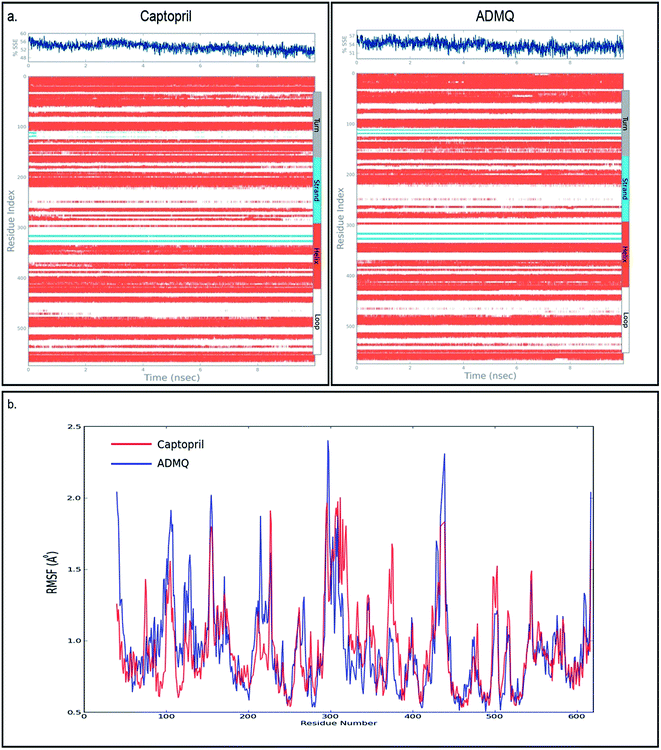 |
| | Fig. 4 (a) The secondary structural elements (SSE) of captopril bound to ACE is shown on top left whereas on top right the secondary structural elements of ADMQ bound to ACE is depicted. (b) Protein RMSF on interaction with antihypertensive drug, captopril (red) and ADMQ (blue). | |
The highly comparable Glide score, 2D ligand interaction diagram, SSE and RMSF clearly shows that both the captopril and ADMQ holds similar interaction mechanism on the enzyme's active site. Hence, the synthesized open chain flavonoid, ADMQ could serve as a potential antihypertensive drug as far as in vitro and in silico results are concerned.
Influence of BSA on ADMQ–ACE binding
To examine the probable influence of carrier protein BSA on the antihypertensive effect of ADMQ, the inhibitory effect on kidney cortex plasma membrane in presence of BSA have been monitored with a full inhibitory profile of ADMQ by extending its concentration range till saturation (Fig. 1a). It is observed that BSA has decreased the antihypertensive activity of ADMQ by a slight extent, which essentially directs that the carrier protein probably is in weak competition with the target enzyme towards ADMQ binding. The similar result has also been observed from the slightly higher inhibitory constant in presence of BSA (207.03 μM) as compared to its absence (167.19 μM) (Fig. 1b).
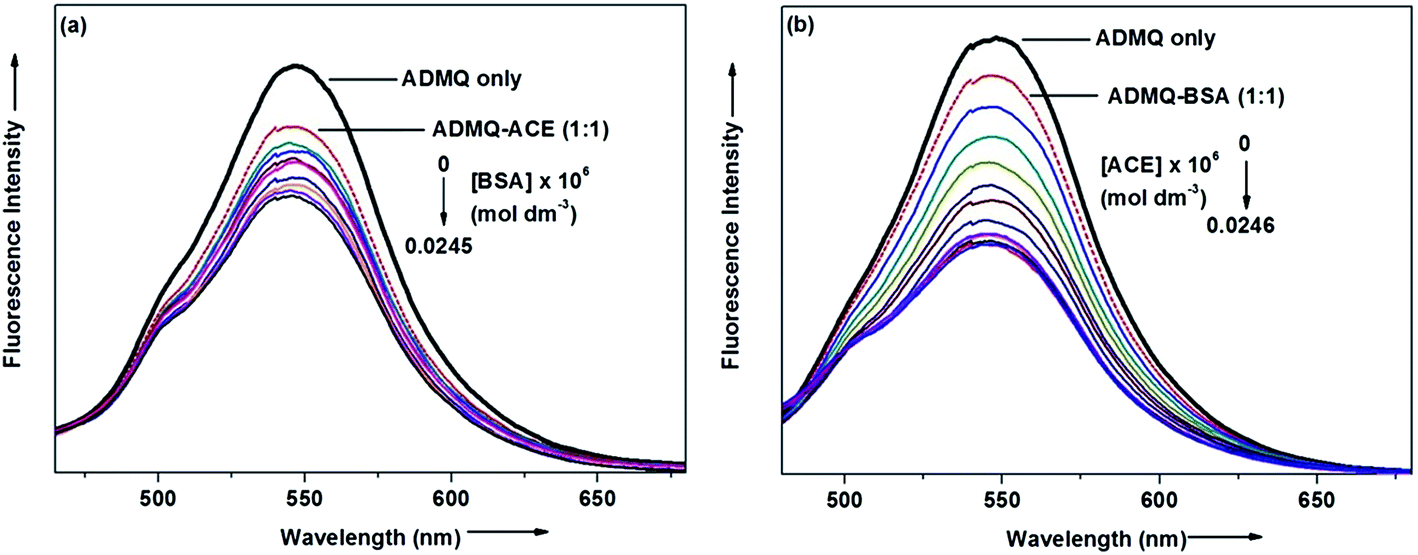
On interaction with carrier protein BSA, ADMQ accommodate itself in a specific manner in the protein scaffold and undergoes a significant binding with BSA (discussed later on). The competitive binding interaction has been experimented in absence and presence of the other interacting partner (BSA/ACE). Both the ACE 1.27 × 109 (±10%) and BSA 2.67 × 108 (±10%) undergoes a comparable binding with ADMQ in absence of each other. But the gradual addition of BSA in the equimolar mixture of ADMQ–ACE system, do not change the emission intensity to a significant extent (Fig. 5a), whereas, in the reverse experiment, ACE is able to efficiently decrease the emission intensity in presence of equimolar concentration of carrier protein (Fig. 5b). This competitive binding interaction is quantified in terms of binding constant Kb (as discussed earlier) and is documented in Table 1. The much lesser ADMQ–BSA binding constant 2.40 × 104 (±18%) in presence of ACE strongly suggests the effective release of the synthesized molecule to ACE. Hence from the above experimental observations it is clear that although there is a weak competition that exists between the ACE and BSA towards ADMQ binding, which essentially decreases its antihypertensive activity by a small extent, but most importantly the carrier protein is unable to hold the ADMQ molecule from the target enzyme. This information could be very useful for the effective delivery of potential antihypertensive drug ADMQ to the target enzyme.
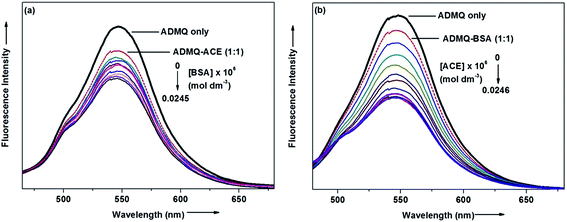 |
| | Fig. 5 (a) Variation in the emission spectra of ADMQ + ACE (1:1) on gradual addition of BSA at 298 K, pH 7.4. (b) Variation in the emission spectra of ADMQ + BSA (1:1) on gradual addition of ACE at 298 K, pH 7.4. | |
Table 1 Binding constants (Kb) of ADMQ with ACE and BSA in absence and presence of each other
| |
In absence of BSA/ACE |
In presence of BSA/ACE |
| Binding constant (Kb) of ADMQ–ACE |
1.27 × 109 (±10%) |
2.57 × 107 (±11%) |
| Binding constant (Kb) of ADMQ–BSA |
2.67 × 108 (±10%) |
2.40 × 104 (±18%) |
Interaction of potential ACE inhibitor ADMQ with carrier protein
The interaction of potential antihypertensive heterocyclic chalcone derivative ADMQ with carrier protein BSA has been investigated to get a clear picture about its delivery mechanism. On gradual addition of ADMQ in aqueous buffer solution of BSA at pH 7.4, the decrement of intrinsic fluorescence intensity has been observed without any appreciable change in the emission maxima at 340 nm (Fig. 6). This progressive diminution in the fluorescence intensity suggests the binding interaction between ADMQ and the carrier protein. The insensitive behavior of the emission maxima on gradual addition of the probe molecule indicates that there is almost no change in the immediate vicinity of tryptophan residues although ADMQ is situated at close proximity (discussed later on) for the interaction to occur. Hence, the observed quenching may be attributed to changes in protein conformation, subunit association and substrate binding.
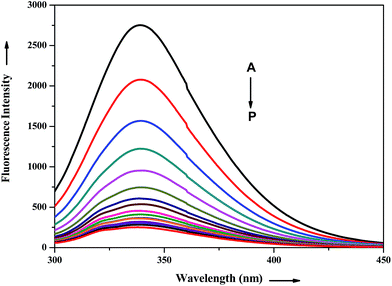 |
| | Fig. 6 Effect of ADMQ on fluorescence spectrum of BSA at 298 K (λex = 280 nm). Curves A → P corresponds to 0, 9.33, 18.57, 27.70, 36.77, 45.74, 54.62, 63.42, 72.13, 80.76, 89.31, 97.11, 106.16, 114.47, 122.70, 130.87 × 10−6 mol dm−3 of ADMQ, respectively. | |
The above fluorescence quenching phenomenon has been quantified by the well-known Stern–Volmer (SV) equation (eqn (2))36,37
| | |
F0/F = 1 + Ksv[Q] = 1 + kqτ[Q]
| (2) |
where
F0 and
F denotes steady state fluorescence intensity of ADMQ–BSA complex in absence and presence of quencher, respectively,
Ksv is the Stern–Volmer quenching constant,
kq is the bimolecular quenching rate constant, [Q] is the total concentration of quencher and
τ is the average lifetime of the protein in the absence of quencher. To study the quenching process carefully, the above said fluorescence quenching experiment has been performed at four different temperatures at 298, 303, 308 and 313 K (
Fig. 7). The calculated Stern–Volmer quenching constants
Ksv and bimolecular quenching rate constant
kq are found to be inversely correlated with temperature as documented in
Table 2. In the case of dynamic quenching, higher temperature results in faster diffusion, and consequently quenching rate constant increases with increasing temperature whereas in the case of static quenching, increasing temperature is likely to result in decreasing complex stability and hence in lowering the value of the quenching constant.
38 Moreover, the estimated
kq value for ADMQ–BSA system at 298 K in the order of 10
12 is much greater than the maximum possible value of the diffusion controlled quenching (
i.e., 2.0 × 10
10 L mol
−1 s
−1).
39 These results suggest a strong interaction between ADMQ and carrier protein
via static quenching mechanism. However, detail time resolved measurements have also been experimented to verify this phenomenon.
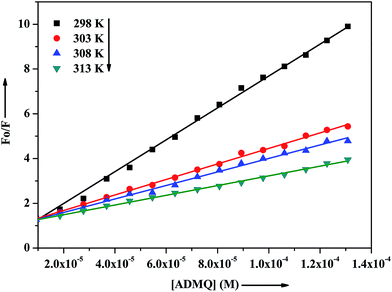 |
| | Fig. 7 Stern–Volmer plots for interaction of ADMQ and BSA at different temperatures. | |
Table 2 Stern–Volmer quenching constants for the interaction of ADMQ with BSA at various temperatures (average lifetime of BSA used is 10−8 s)
| Temp. (K) |
Ksv (104 L mol−1) |
kq (1012 L mol−1 s−1) |
| 298 |
7.173 ± 1.63% |
7.173 ± 1.63% |
| 303 |
3.465 ± 1.23% |
3.465 ± 1.23% |
| 308 |
3.093 ± 3.26% |
3.093 ± 3.26% |
| 313 |
2.169 ± 3.49% |
2.163 ± 3.49% |
Thermodynamic parameters and binding forces
For a complex formation process, modified Stern–Volmer equation (eqn (3)) may be used to calculate the affinity constant Ka between ADMQ and BSA.| | |
F0/ΔF = F0/F0 − F = 1/faKa × 1/[Q] + 1/fa
| (3) |
where fa represents the fraction of initial fluorescence that is accessible to quencher and [Q] is the quencher concentration. The affinity constant Ka at different temperatures is represented in Table 3.
Table 3 Thermodynamic parameters of the system of ADMQ–BSA at different temperatures
| T (K) |
Ka (104 L mol−1) |
Ra |
ΔH (kJ mol−1) |
ΔG (kJ mol−1) |
ΔS (J K−1 mol−1) |
Rb |
SDc |
| R is the correlation coefficient for modified Stern–Volmer plots. R is the correlation coefficient for van't Hoff plots. SD is the standard deviation for van't Hoff plots. |
| 313 |
3.127 |
0.978 |
−16.657 |
−10.069 |
32.119 |
0.997 |
0.195 |
| 308 |
3.194 |
0.954 |
−9.909 |
| 303 |
3.502 |
0.992 |
−9.748 |
| 298 |
4.115 |
0.976 |
−9.588 |
In general, the interactions between small molecules and protein may result mainly from four types of forces: hydrogen bond, van der Waals force, electrostatic interactions and hydrophobic interaction.40 Ross and Subramanian have summed up the thermodynamic laws to determine the type of binding associated with ligand–protein interactions:41 if (1) ΔH > 0 and ΔS > 0, it indicates a dominant hydrophobic interaction; (2) ΔH < 0 and ΔS < 0 suggest that hydrogen bond and van der Waals force are the dominating force and (3) ΔH < 0 or ΔH ≈ 0 and ΔS > 0 imply that electrostatic interactions are dominant. The values of ΔH and ΔS of ADMQ–BSA system has been calculated using van't Hoff equation (eqn (4))
| | |
lnK = −ΔH/RT + ΔS/R
| (4) |
where
K is analogous to the effective quenching constants
Ka at the same temperature and
R is the gas constant. The thermodynamic parameters of ADMQ–BSA system are listed in
Table 3. From the plot of ln
K versus 1/
T (
Fig. 8), the estimated value of Δ
H = −16.657 kJ mol
−1 and Δ
S = 32.119 J mol
−1 K
−1 respectively suggest that the driving force of ADMQ–BSA binding process is mainly electrostatic in nature. However the positive value of Δ
S also indicates that the hydrophobic interactions should not be excluded for the same.
42 The value of Δ
G at different temperatures has also been calculated using
eqn (5).
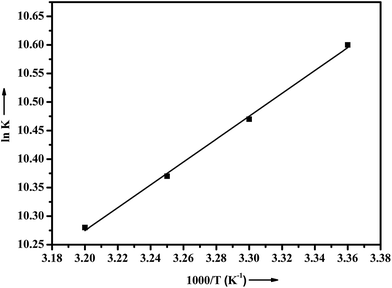 |
| | Fig. 8 Plot of van't Hoff equation of ADMQ and BSA at different temperatures. | |
The negative sign for free energy (ΔG) of the ADMQ–BSA system (Table 2) indicates spontaneous interaction between these two.
Protein induced motional restriction
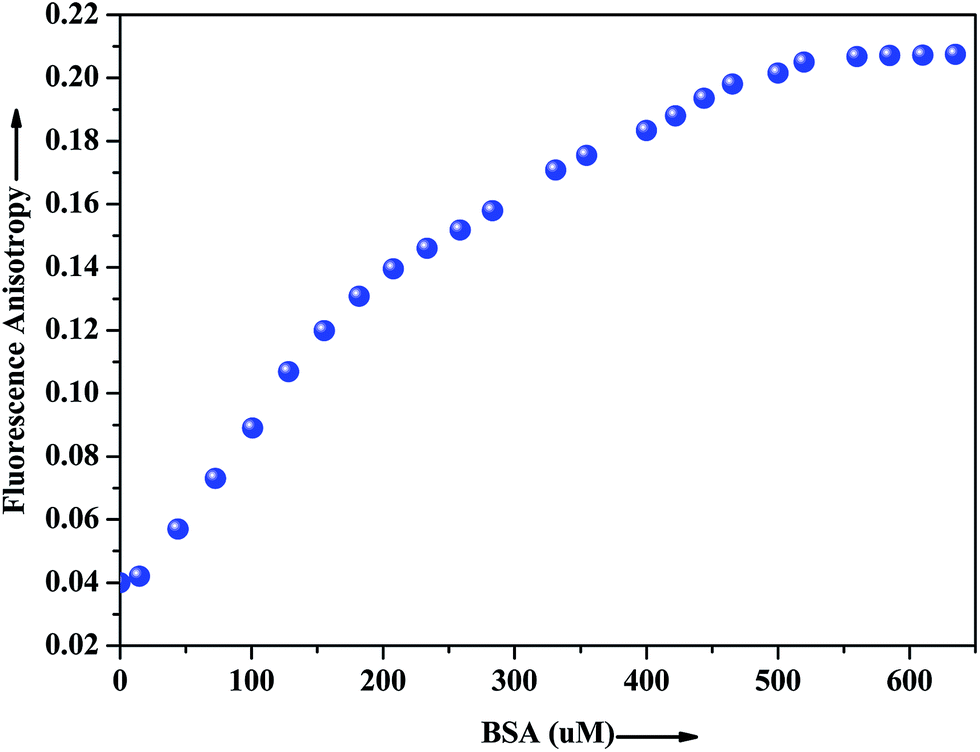
The extent of restriction imposed by the microenvironment on the dynamic properties of the probe can be exploited in assessing the motional information in the microheterogeneous environments of proteins. In this regard, anisotropy measurement has been employed to gather additional evidence in support of the interaction of the probe with the native albumin. In fluids, the anisotropy value is very low because of the free rotation of the fluorophore, whereas in restricted environment like in protein, DNA, micelles, microemulsion etc. the anisotropy values goes up.43 The fluorescence anisotropy monitored for ADMQ shows a marked increase on moving from aqueous phase to the protein environments, revealing that the rotational diffusion of the probe molecule is restricted significantly. Fig. 9 presents the variation of the fluorescence anisotropy (r) of the probe molecule monitored at 550 nm as a function of protein concentration. The plot reveals that with increasing protein concentrations, up till 50 × 10−5 mol L−1 fluorescence anisotropy (r) increases rapidly and then levels off gradually (r = 0.206). The gradual increase in fluorescence anisotropy of ADMQ with an increasing protein concentration implies an imposed motional restriction on the fluorophore in the proteinaceous environments which indicates that the emission is literally coming from entrapped ADMQ anywhere in the binding domain of BSA.
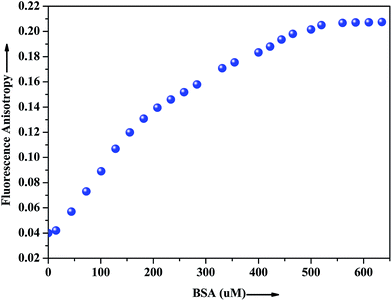 |
| | Fig. 9 Variation of fluorescence anisotropy (r) of ADMQ with increasing concentration of BSA. λex = 430 nm for ADMQ. λem = 550 nm. | |
Identification of binding domain on carrier protein
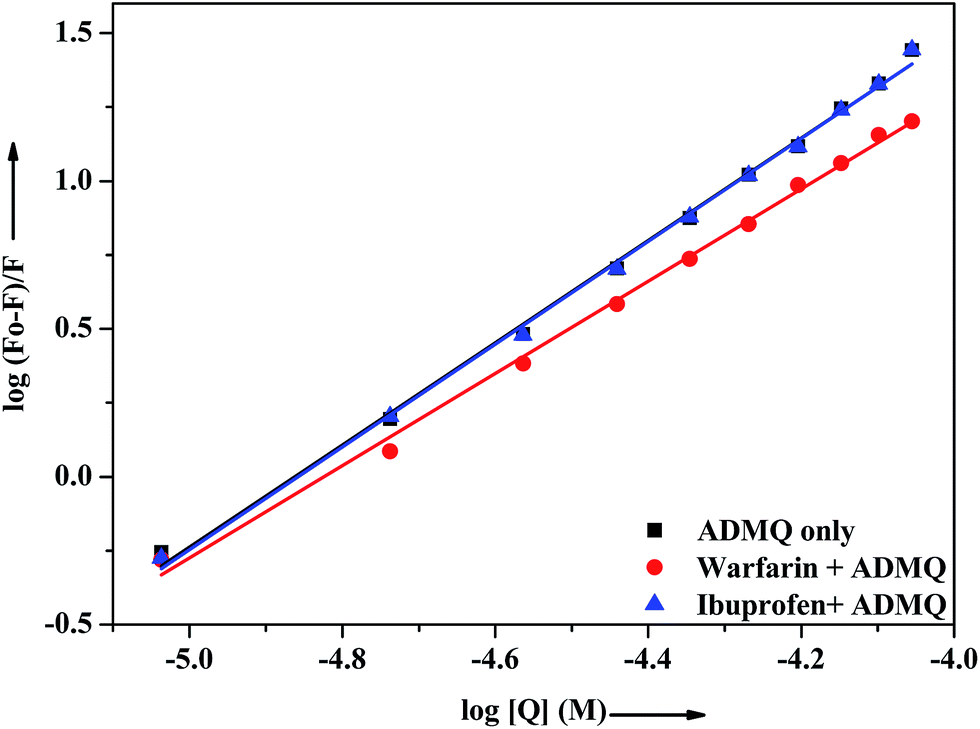
In vitro site specific interaction studies. To ascertain the precise ADMQ binding location on BSA, competitive binding experiments have been carried out to clarify its binding properties,22,44 using probes that specifically bind to the known site or domain. Sudlow et al.45 have suggested two distinct binding sites on BSA, namely site I and site II. The flexibility of site I is such that it can easily accommodate a large number of ligands such as bicarboxylic acids, bulky negatively charged heterocycles, long chain fatty acids, indole derivatives and aldehydes like warfarin, phenylbutazone by binding to them.46 Another relevant binding site is known as ibuprofen binding site and it is located in site II (subdomain IIIA).47 The interior of the cavity in site II is constituted of hydrophobic amino acid residues, and the pocket exterior presented two important amino acid residues (Arg 410 and Tyr 411).48 This site binds preferentially aromatic carboxylic acids though it exhibits a high selectivity also for anions of long chain fatty acids (C8).On addition of warfarin into BSA solution, the Trp emission intensity at 340 nm decreased significantly and on gradual addition of the probe ADMQ into it, the fluorescence intensity of protein–warfarin complex (1:1) decreases further. The similar observation has also been noticed when the probe ADMQ is added in ibuprofen–BSA complex (1:1). However the quenching efficiency is different in each case. The binding interaction of ADMQ–BSA system in absence/presence of site markers (warfarin/ibuprofen) has been calculated using eqn (1). The binding constants (Kb) for ADMQ–BSA alone (without any site marker) is found to be 2.55 × 108 (±10%) at 298 K and pH 7.4, whereas the binding constant of ADMQ in presence of warfarin and ibuprofen (at 298 K and pH 7.4) are found to be 3.32 × 107 (±8%) and 2.79 × 108 (±10%) respectively (Fig. 10). Similar magnitude of binding constants for ADMQ–BSA complex in presence and absence of ibuprofen indicates that site II marker ibuprofen does not compete with ADMQ at its binding site. The decrease binding constant (Kb) value in presence of warfarin (3.32 × 107) implies that ADMQ competes with warfarin while binding to the site I of the protein. Hence the above site specific control experiments suggest that ADMQ binds preferentially to the ‘warfarin binding site’ i.e. subdomain IIA of site I of the macromolecule.
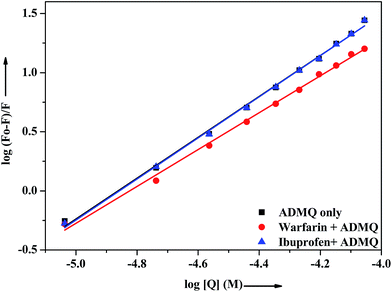 |
| | Fig. 10 Effect of selected site markers on ADMQ–BSA system in 0.05 M HEPES buffer of pH 7.4 at 298 K. [BSA]:[site marker] = 1:1. | |
Putative binding site analysis by in silico approach
To corroborate the in vitro spectroscopic findings, the site specific interaction between BSA and ADMQ has been investigated by employing a simulation strategy that combines molecular docking, induced fit docking and molecular dynamics simulation in a rational workflow.
Molecular docking study
The mode and orientation of small molecules attached to the serum albumin have profound influence on their bioactivity in physiological conditions.49 The tertiary structure of BSA has been resolved through X-ray crystallography50 and thereby offers the basis of the localization pattern of any ligand/drug molecule into it. Crystallographic data has shown that the binding regions for various ligands are generally localized within subdomain IIA or IIIA of the protein molecule; according to Sudlow's nomenclature, these binding domains correspond to sites I and II, respectively.
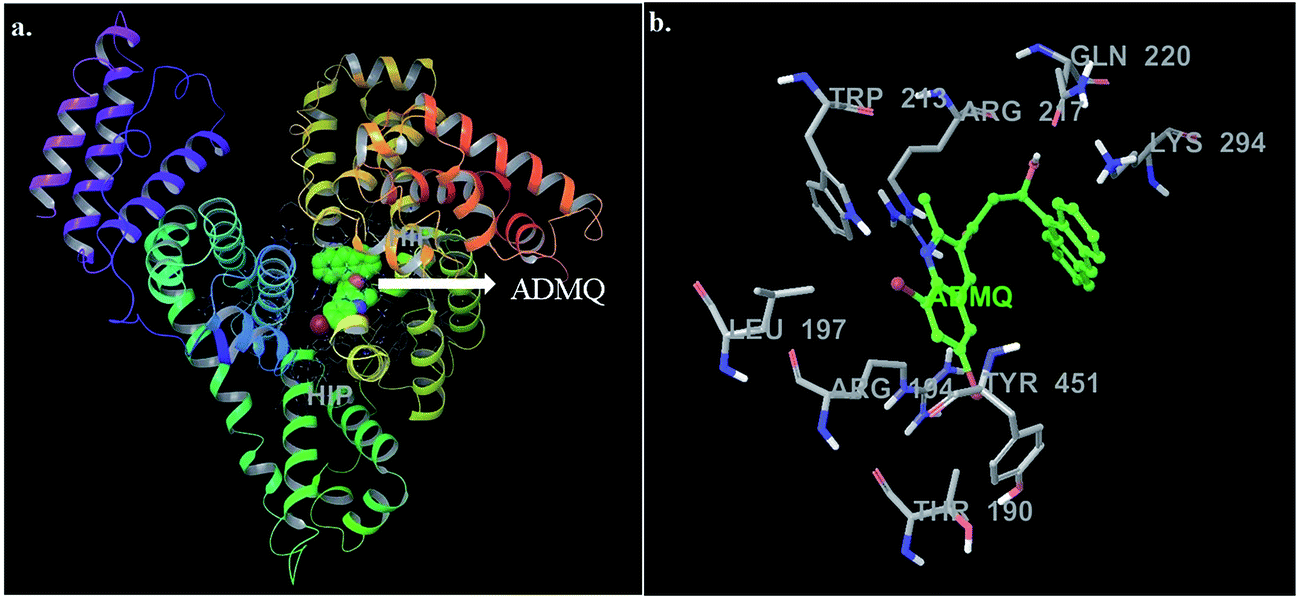
In this context, molecular docking studies has been carried out to assess the binding site of carrier protein chosen by β-hydroxy form of ADMQ (since this is expected to be present in higher extent in physiological environment),29 its preferred orientation and detailed mechanism of interaction using Glide programme.51 Molecular docking studies suggest that the energetically most favorable binding site of the probe is located inside a hydrophobic cavity of subdomain II A or Sudlow site-I, near Trp 213 amino acid residue of BSA (Fig. 11a). This first binding site (BSI), also called as “Warfarin binding site”, is constituted by a hydrophobic pocket surrounded by positively charged amino acids. In docking studies, the hydrophobic and H-bonding interactions play a major role for the binding and stabilization of ADMQ snugly fitted into subdomain IIA. Hydrophobic interactions of ADMQ with nearby protein residues Trp 213, Leu 197, Val 342, Tyr 451, Leu 454, Tyr 340, and Ala 341 facilitate the anchoring of the probe inside the protein pocket, while residues Gln 220 and Thr 190 make polar contacts with the probe in the BSA–ADMQ complex (Fig. 11b). The comparable dock score (−5.70 kcal mol−1) and MM-GBSA binding energy (−76.48 kcal mol−1) of ADMQ with that of warfarin (Table 4) signifies that ADMQ is stable while sitting in BS I protein scaffolds and this in silico result is in line with the site-specific experimental observation.
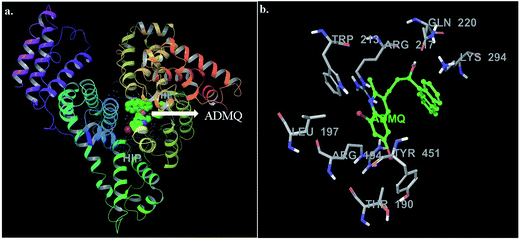 |
| | Fig. 11 (a) Minimum energy conformation of ADMQ inside the scaffold of IIA subdomain of BSA (3V03). (b) A close view of amino acid residues in the immediate vicinity of ADMQ. | |
Table 4 Energetics of ADMQ and warfarin site on site I of BSA
| Title |
Docking score (kcal mol−1) |
Glide Emodel |
MM-GBSA (kcal mol−1) |
| Warfarin |
−6.95 |
−44.91 |
−63.18 |
| ADMQ |
−5.70 |
−41.37 |
−76.48 |
Induced fit docking study
In order to provide a clear understanding of the binding affinity of the ligand ADMQ with BSA, induced fit docking (IFD),52 which is capable of sampling dramatic side chain conformational changes as well as minor changes in backbone structure has been conducted using Prime program in Schrödinger's IFD module53 for ADMQ–BSA system. It consists of soft-docking stage in which ADMQ is docked into a structural ensemble of receptor conformations, followed by a complex minimization stage for the highest ranked poses, during which both the ligand and the receptor binding site are free to move.
For ADMQ–BSA system, the IFD protocol shows slight structural perturbations in the receptor site (BSA) where ADMQ binds deep within the hydrophobic pocket in its minimum energy conformation. ADMQ acquires an arc shape conformer and possesses an angle of 113.1°. It is also noticed that the quinoline moiety of ADMQ lies in close vicinity of Trp 213 and the average distance between the two moieties has been estimated to vary between 0.37 nm and 1.45 nm. The distance between tryptophan and the bound probe (2.45 nm) obtained from FRET (discussed later) is also comparable with the distances obtained from induced fit docking (Fig. 12a). For ADMQ–BSA system, the Glide and IFD score of −5.70 kcal mol−1 and −5891.13 kcal mol−1 respectively suggest a high protein binding affinity of ADMQ in aqueous buffer environment at pH 7.4. 2D ligand interaction diagram (Fig. 12b) suggests that the residues in the immediate vicinity of the probe molecule are majorly hydrophobic side chains (Trp 213, Leu 197, Leu 454, Tyr 451, Tyr 340) and also the positively charged residues, namely Arg 194, Arg 217, Lys 294 and Lys 221. Within a distance of 1.89 Å, one hydrogen bond is also present between the hydroxyl group of ADMQ and polar amino acid Gln 220. Hence hydrophobic, electrostatic and hydrogen bonding interactions are all involved in securing the ligand into the protein.
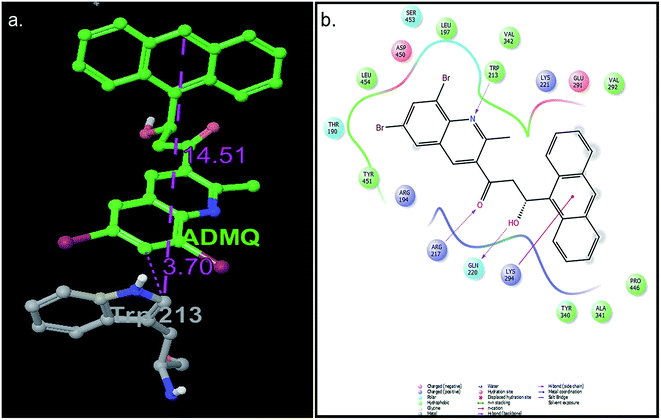 |
| | Fig. 12 (a) Distance between bound ADMQ and Trp 213 in the minimum energy docking pose presented by ball and stick model, distance shown is in Å. (b) 2D ligand interaction diagram of amino acid residues in the immediate vicinity of ADMQ. | |
Molecular dynamics simulation
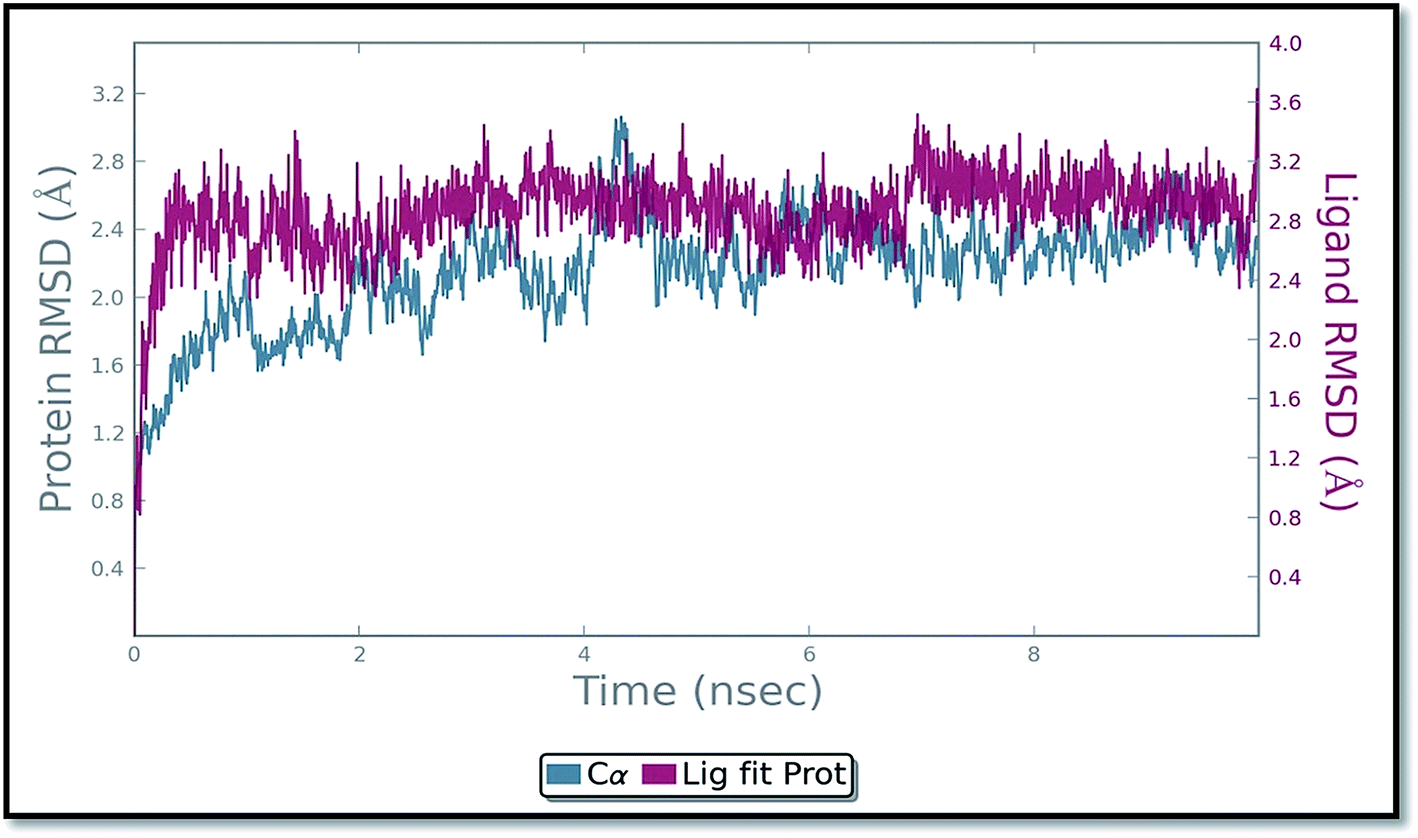
The molecular dynamics simulation (MD) has also been carried out in addition with regular XP docking and IFD study to determine the main features of ADMQ–BSA interaction especially when both protein and ligand are free to move in presence of solvent. The best pose of ADMQ–BSA conjugated system generated by IFD study is considered for the explicit real time MD simulation for 10 ns by Desmond,54 where system builder use explicit aqueous medium followed by complex minimization to its lowest energy level. In MD simulation the stability of ligand ADMQ in the proteinaceous environment is represented in terms of Root Mean Square Deviation (RMSD). The protein and ligand RMSD for BSA–ADMQ system is depicted in Fig. 13. This ‘Lig fit Prot’ shows the RMSD of the ligand when the protein–ligand complex is first aligned on the protein backbone of the reference and the RMSD of the ligand heavy atoms is measured. This depicts that ADMQ initially explores the surrounding of the docking placement while remaining in contact with the protein chain and it has not been diffused away from the initial protein binding site. The change in RMSD of the protein to the order of 1–3 Å during entire run indicates that the protein has undergone minor conformational change during the simulation.
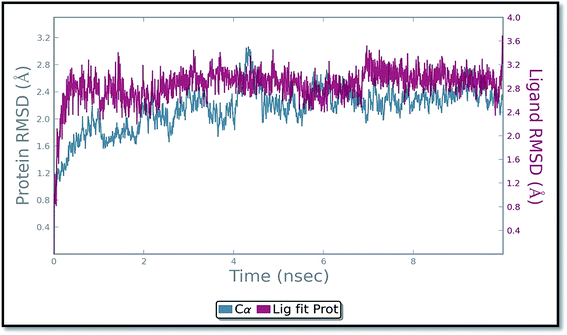 |
| | Fig. 13 Protein and ligand (ADMQ) RMSD plot during 10 ns simulation. | |
Conformational change of BSA upon ADMQ binding
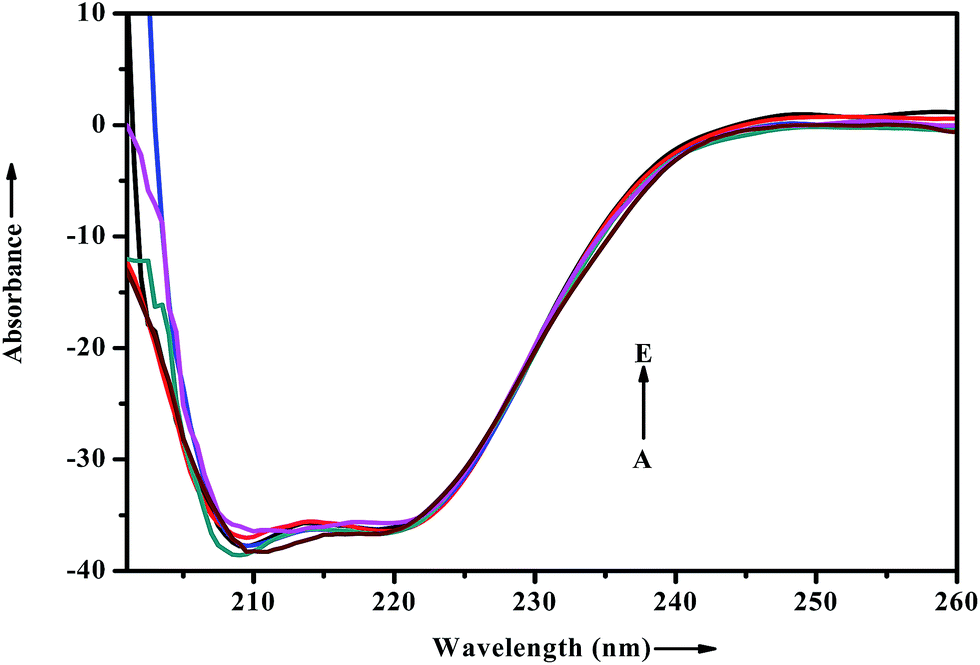
When a drug/probe molecule binds to the carrier protein, it may cause conformational changes in the tertiary structure of proteins. The transportation mechanism of drug depends on the conformational ductility and adjustability of the protein (host) to facilitate the interaction with the drug (guest) molecule. This conformational change may affect the biological function of serum albumins as the carrier protein. Circular dichroism is often used to quantitatively analyze the secondary structural changes of protein (in terms of α-helices, β-sheets, turn and random coil and the possible interconversions) induced by the binding interaction with small molecules.55 The structural characterization of proteins depends greatly on the remarkable sensitivity of CD in far-UV region. To ascertain the possible influence of ADMQ binding on the secondary structure of BSA, CD measurements is performed in the range of 200–260 nm in presence of different ADMQ concentrations at pH 7.4 at room temperature (Fig. 14). The CD spectra of BSA exhibits two negative bands in the far-ultraviolet region at about 208 (π–π*) and 222 nm (n–π*), which is characteristic of the alpha conformation of protein.56,57 The CD spectra shows that the protein structure after probe binding is predominantly α-helical. The regular decrease in the intensity of negative CD band at 208 nm on gradual addition of probe molecule (Fig. 14) reveals minor reduction in the α-helical content. The α-helical content in free BSA is found to be 79.60% under the experimental condition and this is reasonable with the available literature.58 A small decrease in the α-helix content from 79.60% to 77% suggests minor peptide strand unfolding and it indicates that ADMQ has little effect on the conformation of BSA. Thus, the binding of ADMQ may have changed the atomic arrangement of the amino acid residue in BSA to a minor extent and partially destroyed its hydrogen bonding network and therefore the polypeptide chain becomes tender to accommodate the probe molecule in a specific manner inside the protein scaffold. Hence, this minor decrease in the alpha helical content may not affect the physiological function of this carrier protein during transportation of this antihypertensive ADMQ molecule.
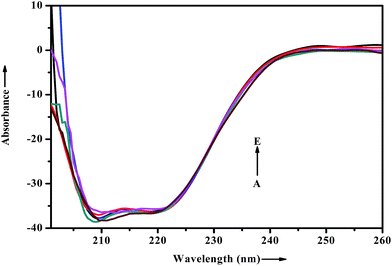 |
| | Fig. 14 Effect of increasing ADMQ on the CD spectra of BSA. [BSA] = 2.4 × 10−6 mol dm−3; ADMQ, A → E: 3.73, 7.45, 22.18, 40.27 and 58.01 × 10−6 mol dm−3. | |
Investigation of conformational change by synchronous fluorescence
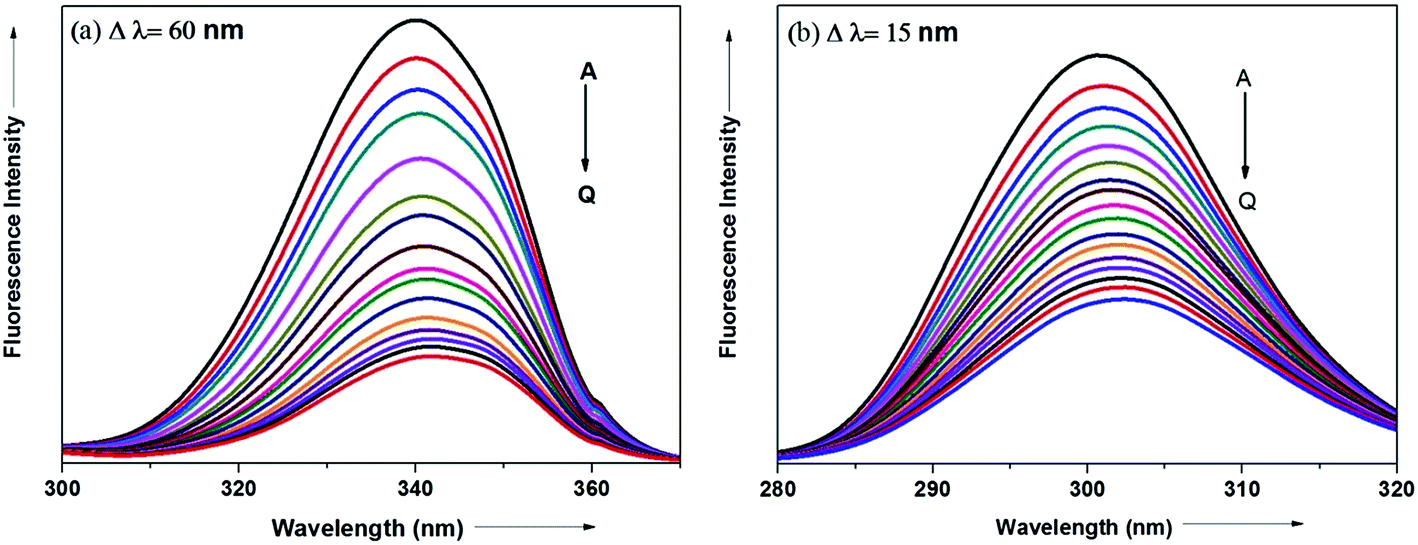
The synchronous fluorescence spectrum is a useful method to characterize the interaction between fluorescence probe and protein because it can provide information about molecular microenvironment in the vicinity of fluorophore.57,59 According to Miller et al., when the scanning interval (Δλ) between emission wavelength (λem) and excitation wavelength (λex) is stabilized at 60 and 15 nm, the synchronous fluorescence gives the characteristic information of Trp and Tyr residues of protein, respectively.55,60 The maximum emission wavelengths (λmaxem) of tryptophan and tyrosine residues in the protein molecule are related to the polarity of their surroundings whereas the shift in the λmaxem correspond to changes in protein conformation. Fig. 15a and b displays the effect of addition of antihypertensive ADMQ with varied concentrations on the synchronous fluorescence spectra of BSA when Δλ are 60 and 15 nm respectively.
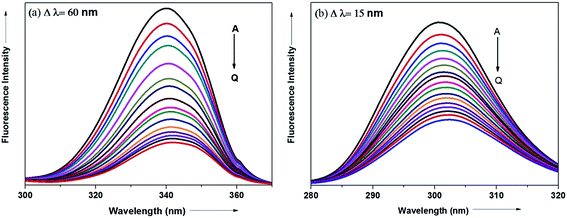 |
| | Fig. 15 Synchronous fluorescence spectra of BSA in the presence of different concentrations of ADMQ. [BSA] = 5 μM; curves A → Q corresponds to 0, 1.24, 2.48, 3.71, 4.95, 6.18, 7.40, 8.63, 9.85, 11.0, 12.2, 13.4, 14.7, 15.9, 17.1, 18.3, 19.5 × 10−6 mol dm−3 of ADMQ respectively at 298 K; pH = 7.4 (a) Δλ = 60 nm; (b) Δλ = 15 nm. | |
In the above experiment, both the Trp and Tyr emission maxima shows a minor 2 nm red shift at the investigated concentration range which could be due to the minor conformational alteration in the protein chain.60,61 This observation is in line with the CD results where ADMQ exhibited minor perturbation in the alpha helical content of BSA. This tiny bathochromic shift also signifies that the hydrophobicity around fluorophore is slightly decreased and both the Trp and Tyr residues are marginally moved from nonpolar hydrophobic environment within the protein cavity to a slightly polar hydrophilic environment after interaction with ADMQ. This is also supported by thermodynamic parameters where the driving force of ADMQ–BSA binding is predominantly electrostatic in nature.
In addition, the decrease in the fluorescence intensity of Trp synchronous fluorescence is somewhat higher (75%) as compared to Tyr (60%). This implies that the binding region is possibly located a bit closer to the Trp residue rather than Tyr residue of protein,57 concerning Trp residue being implicated more in the association process with the ADMQ molecule.
Modulation of excited state dynamics
Lifetime measurements have been carried out to verify the possibility of involvement of simultaneous dynamic process in quenching of BSA fluorescence by ligand. Albeit static and dynamic quenching may be differentiated by their diverging dependence on temperature and viscosity,36 fluorescence quenching is preferably acquired by time-resolved fluorescence (TRF) measurements, which can distinguish between static and dynamic process immediately.
There are two lifetime components obtained under the experimental condition for native BSA: one (τ1 = 2.54 ± 0.08 ns) contributing 35% of total fluorescence and the other (τ2 = 6.64 ± 0.03 ns) contributing the rest of the fluorescence which is in line with the reported literature.62 The one with a longer lifetime is assigned to Trp 134, and the other is assigned to Trp 213.63 The relative contribution of each component is dependent on the folding/unfolding conditions due to the protein's multiple local configurations and changes in the extend of solvent accessibility. Thus, the information of the protein conformational behavior can be obtained from the emission spectra of tryptophan in ADMQ–BSA bioconjugate.
The fluorescence lifetime decay profiles of native BSA and BSA–ADMQ composites in HEPES buffer, pH 7.4, are represented in Fig. S3.† The amplitudes along with the various statistical parameters used to check the goodness of fit are pooled in Table 5. The best fits for BSA are obtained using a biexponential function; which is typical for tryptophan in proteins.62,64 On addition of ADMQ the lifetime value of Trp 213 residue in BSA (τ1) is observed to decrease prominently whereas the lifetime (τ2) of Trp 134 shows insignificant change. This result practically supports our earlier observations of site-specific binding of ADMQ in BS I (where Trp 213 resides) as well as in silico IFD results.
Table 5 Lifetime data of BSA in ADMQ environment at 298 K
| [ADMQ] (μM) |
τ1 (ns) |
τ2 (ns) |
α1 |
α2 |
τm (ns) |
χ2 |
| 0 |
2.54 |
6.64 |
0.29 |
0.53 |
5.19 |
1.20 |
| 9.33 |
2.34 |
6.68 |
0.31 |
0.51 |
5.05 |
1.14 |
| 18.56 |
1.91 |
6.57 |
0.33 |
0.53 |
4.78 |
1.22 |
| 27.71 |
1.79 |
6.55 |
0.37 |
0.50 |
4.54 |
1.31 |
| 36.77 |
1.55 |
6.46 |
0.41 |
0.50 |
4.24 |
1.10 |
| 45.74 |
1.13 |
6.33 |
0.55 |
0.46 |
3.50 |
1.15 |
| 54.62 |
0.78 |
6.18 |
0.75 |
0.44 |
2.77 |
1.22 |
| 63.42 |
0.61 |
5.99 |
0.94 |
0.39 |
2.18 |
1.20 |
| 72.13 |
0.50 |
5.94 |
1.23 |
0.34 |
1.67 |
1.17 |
| 89.31 |
0.32 |
5.73 |
1.94 |
0.26 |
0.97 |
1.16 |
| 122.70 |
0.18 |
5.49 |
2.69 |
0.16 |
0.43 |
1.13 |
The mean fluorescence lifetime (τm) is also been monitored and calculated using the following equation65
where
αi is the initial intensity of the decay component
i, having a lifetime
τi. This equation serves as an important parameter for exploiting the behavior of protein molecule bound to ADMQ. The mean lifetime of BSA (
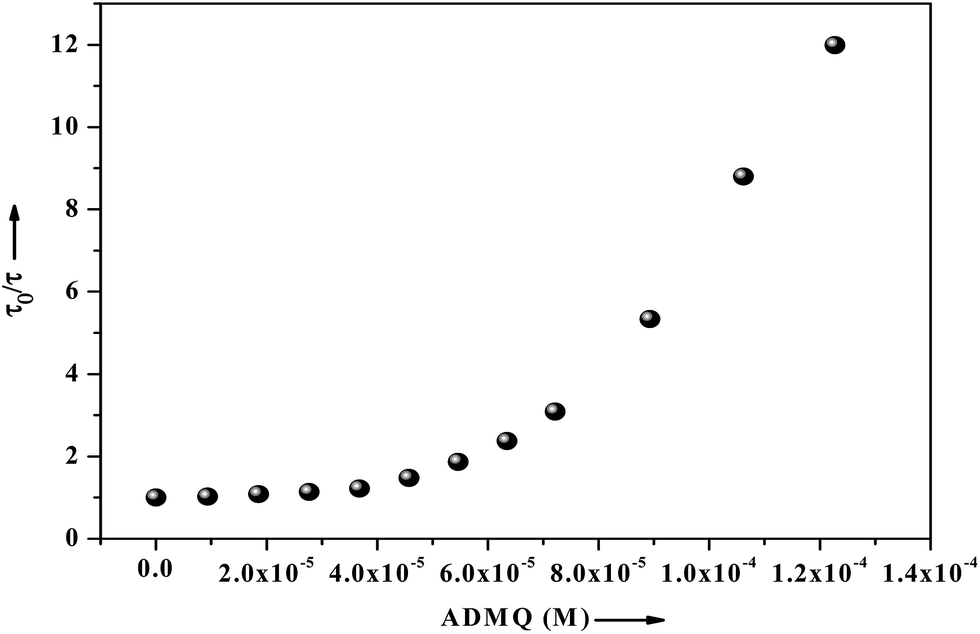
τm) significantly decrease from 5.19 ns (in buffer) to 0.43 ns with the gradual increment of ADMQ concentration. The Stern–Volmer plot obtained from the mean fluorescence lifetimes is shown in
Fig. 16, which is linear up to ADMQ concentration of 30 μM, but interestingly it exhibits an upward concave deviation towards the
y-axis at higher concentration of the probe molecule. This observed positive deviation from linearity indicates several possibilities like (a) the presence of a sphere of action
i.e., the existence of a sphere of volume around a fluorophore within which a quencher being adjacent to the fluorophore at the moment of excitation, will cause a quenching with probability of unity;
66 (b) the existence of combined quenching (static and dynamic) processes simultaneously as a result of energy transfer between the fluorophore and the complex formed; or most significantly (c) the existence of more than one binding site,
i.e. a model which involves two kinds of fluorophores
viz. Trp 213 and Trp 134; one which is available for quenching (Trp 213) by ADMQ and the other (Trp 134), which is not.
67 This may results a biphasic nature SV plot. However above three possibilities may be operative independently or simultaneously with one another.
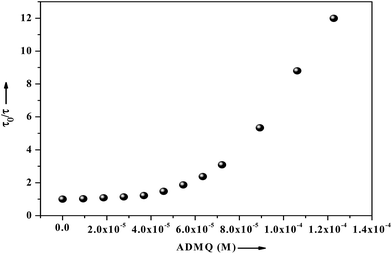 |
| | Fig. 16 Fluorescence lifetime of BSA (2.6 μM) as a function of ligand concentration ranging from 0 to 122 μM, where τ0 and τ are the mean lifetime of BSA in the absence and presence of ADMQ. | |
Energy transfer between ADMQ and SAs
To enquire of the involvement of the dynamic process the possibility of Fluorescence Resonance Energy Transfer (FRET) from protein to ligand has been verified for ADMQ–BSA system. According to Forster's theory, FRET efficiency depends mainly on the following factors: (i) a substantial overlap between the emission of the donor and absorption of the acceptor, (ii) proper orientations of the donor and acceptor transmission dipoles, and (iii) the distance between the donor and acceptor needs to be 2 to 8 nm.68
The normalized respective absorption and emission spectrum of ADMQ and BSA is shown in Fig. S4† which shows a substantial overlap between these two. The efficiency of the possible energy transfer and distance between the donor (BSA) and acceptor (ADMQ) is calculated using Forster equation69
| | |
E = 1 − F/F0 = Ro6/Ro6 + r6
| (7) |
where
E denotes the efficiency of energy transfer between the donor and acceptor and
r is the distance between the donor and acceptor. The critical distance
Ro at which the transfer efficiency equals 50%, is calculated by the following equation:
| | |
Ro6 = 8.79 × 10−25 × [K2n−4ɸDJ(λ)]
| (8) |
where
K2 is the orientation factor to the donor and acceptor of dipoles and is equal to 2/3 for random orientation as in fluid solution,
n is the refractive index of the medium,
ɸD is the fluorescence quantum yield of the donor, and
J expresses the degree of spectral overlap between the donor emission and acceptor absorption, which is obtained from the equation
| | |
J(λ) = ∫Fd(λ)εa(λ)λ4dλ/∫Fd(λ)dλ
| (9) |
where
Fd(
λ) is the normalized donor emission spectrum in the range from
λ to
λ + Δ
λ and
εa(
λ) is the molar absorption coefficient of the acceptor at wavelength
λ. In the present case, we took
εa = 7.487 × 10
3 M
−1 cm
−1 at 430 nm,
n = 1.37 and
ɸ = 0.15. Using
eqn (7)–(9), the following data are obtained:
J = 4.88 × 10
13 M
−1 cm
−1 nm
4,
Ro = 2.223 nm,
E = 0.3574 and
r = 2.45 nm. From this results, it is evident that the values for r is in the 2–8 nm scale, and the acquisition of 0.5
Ro <
r < 1.5
Ro, means that the calculated results are well predicted by Forster's theory demonstrating an existence of an energy transfer between ADMQ and BSA.
Protein aggregation analysis
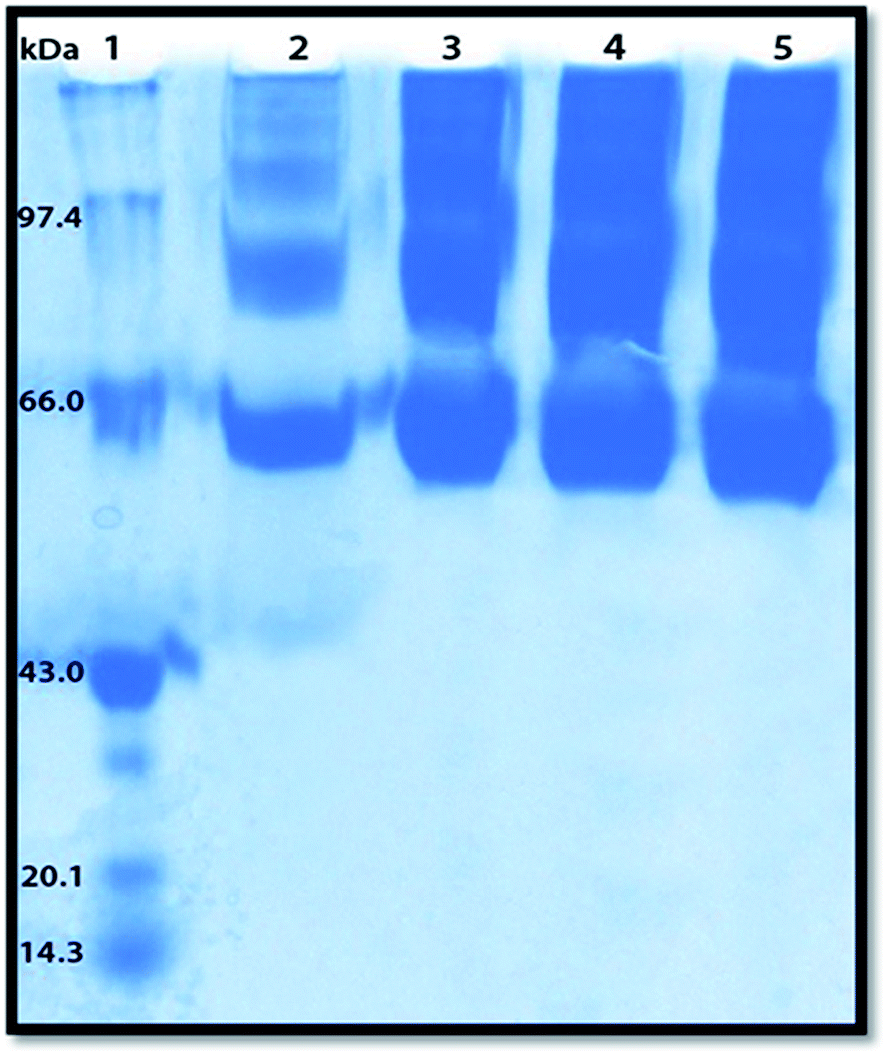
Polyacrylamide gel electrophoresis (PAGE) has been used to visualize the potential protein aggregation resulting from binding of probe to serum albumin.70 Polyacrylamide gel electrophoresis (PAGE), describes a technique widely used in biochemistry, forensics, genetics, molecular biology and biotechnology to separate biological macromolecules, usually proteins, according to their electrophoretic mobility.
In this experiment the migration pattern of BSA treated with ADMQ is different with respect to untreated protein as observed in SDS PAGE. BSA–ADMQ bands became broader than untreated BSA (Fig. 17) after an incubation period of 12 h which indicates the probe ADMQ binds with BSA to form an aggregate. Hence the polyacrylamide gel electrophoresis has also confirmed the formation of BSA–ADMQ conjugate. This behavior is also consistent with our previous experimental and in silico observations.
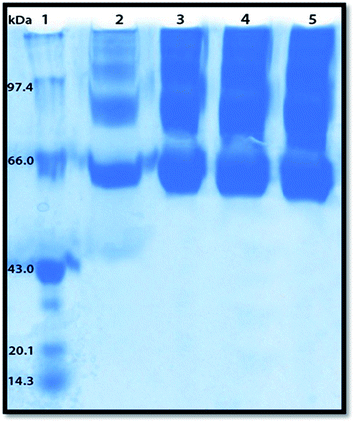 |
| | Fig. 17 Sodium dodecyl sulphate-polyacrylamide gel electrophoresis pattern in 10% gel stained with Coomassie brilliant blue for proteins. Lane 1, molecular weight standard markers (phosphorylase B, 97.4 kDa; bovine serum albumin (BSA), 66 kDa; ovalbumin, 43 kDa; soyabean trypsin inhibitor, 20.1 kDa; lysozyme, 14.3 kDa); Lane 2, native BSA; Lanes 3–5, BSA with increasing ADMQ concentration after an incubation period of 12 h. | |
Experimental section
Materials and methods
ADMQ has been synthesized as reported elsewhere.29 Angiotensin Converting Enzyme (ACE) and captopril has been procured from Sigma Aldrich (USA). Bovine serum albumin (BSA) and N-[2-hydroxyethyl]piperazine N-[2-ethanesulphonic acid] (HEPES) buffer was purchased from SRL, India. Kidney cortex plasma membrane was used as ACE. Stock solution of BSA was prepared by dissolving solid material in 0.05 M HEPES buffer (pH = 7.4) and stored at 4 °C. The same solution was used in all studies of ADMQ in protein. Ibuprofen and warfarin were procured from Bangalore fine chemicals, India. Stock solution of BSA and warfarin were prepared by dissolving them in 0.05 M HEPES buffer (pH = 7.4) and stored at 4 °C. Concentrated stock solution of ADMQ and ibuprofen were prepared in spectroscopic grade 1,4-dioxane. Millipore Water was used to prepare solutions wherever required. Appropriate blanks, run under same conditions, were subtracted from the sample spectra. Water used in all procedures was prepared using a Millipore water purification system.
For antihypertensive activity of ADMQ, the compound was tested at eight concentrations 1, 5, 10, 15, 20, 25, 30 and 35 μg/100 μl dissolved in assay buffer (10 mM HEPES buffer containing 0.3 M NaCl and 10 μM zinc sulphate) containing kidney cortex plasma membrane (ACE enzyme source) and 1 mM Hippuryl-His-Leu as substrate. Another set of ADMQ samples mixed with equimolar concentrations of BSA were also assayed for inhibitory activity of ACE. The compounds (ADMQ) and ADMQ + BSA (1:1) were incubated with enzyme for 10 min at 37 °C. Substrate is then added and incubated for 45 min at 37 °C. The reaction is terminated by the addition of 1 M HCl. The yellow colour is developed by addition of 100 μl of pyridine and 50 μl of benzene sulphonyl chloride. The yellow colour that formed is measured at 410 nm in an ELISA plate reader.71
Instrumentation
Steady-state spectral measurements. UV-Vis absorption spectra were acquired from JASCO (model V-630) spectrophotometer with matched pair quartz cuvettes. The steady state fluorescence emission and excitation spectra were recorded on JASCO FP-8300 with slit width of 2.5 nm. An excitation wavelength of 280 nm was chosen and appropriate blanks corresponding to the buffer solution were subtracted to correct the background. Spectral background corrections were done with similar set of solutions with omission of the probe from within. All measurements were done repeatedly and reproducible results were obtained.The fluorescence of BSA was corrected for the inner filter effect at excitation and emission wavelengths, using the following equation.57,58,72 Appropriate blanks were subtracted from the corresponding data.
| Fcorr = Fobs × antilog[Aex + Aem/2] |
where
Fcorr and
Fobs are the corrected and observed fluorescence intensities, respectively, and
Aex and
Aem are the absorbance at the excitation and emission wavelengths, respectively.
Measurement of steady state fluorescence anisotropy was made on same spectrofluorometer with the parallel and perpendicular emission polarizers, controlled manually. Fluorescence anisotropy (r) is defined as
| r = (Ex90, Em90) − {G(Ex90, Em0)}/(Ex90, Em90) + {2G(Ex90, Em0)} |
where Ex
90 and Ex
0 denotes excitation polarizers in vertical and horizontal positions respectively and Em
90 and Em
0 are emission polarizers oriented vertically and horizontally respectively.
The G value is calculated using formula
| G = (Ex0, Em90)/(Ex0, Em0) |
Circular dichroism spectroscopic measurement. Circular dichroism (CD) spectra were recorded on JASCO J-815 spectropolarimeter at 298 K using a rectangular quartz cuvette of path length 1 cm. The reported CD profiles were an average of four successive scans with 20 nm per minute scan and an appropriately corrected baseline over a wavelength range of 260–200 nm. Each spectrum was baseline corrected and the final plot was taken as an average of three accumulated plots. The content of the α-helix was calculated, and the differential was calculated from CD spectra. The molar ellipticity [θ] was calculated from the observed ellipticity θ as,
where c is the concentration of the protein solution in mol dm−3 and l is the path length of the cell in cm. The CD results were expressed in terms of mean residual ellipticity (MRE) in degree cm2 per dmol according to the following equation:
| MRE = observed CD (mdegree)/cpnl × 10 |
where cp is the molar concentration of the protein, n is the number of amino acid residues (585), and l is the path length (1 cm). The α-helix contents of the free and combined BSA are calculated from the MRE value at 208 nm using the following equation:
| α-helix (%) = [−MRE208 − 4000/33000 − 4000] × 100 |
where MRE208 is the observed MRE value at 208 nm, 4000 is the MRE of the β-form and random coil conformation cross at 208 nm, and 33000 is the MRE value of a pure α-helix at 208 nm.
Synchronous fluorescence spectroscopy. Synchronous fluorescence spectra of BSA in the absence and presence of increasing amounts of ADMQ were measured under the same condition with steady state fluorescence. The spectra of protein samples were obtained after scanning them in the wavelength range of 280–320 nm and 300–370 nm for the difference between excitation and emission wavelengths (Δλ) of 15 and 60 nm, respectively.
Time resolved spectral measurements. Time resolved fluorescence measurements were performed from time resolved intensity decay by a method of time correlated single photon counting (TCSPC) on PTI Fluorescence Picomaster using a pulse diode excitation source at 280 nm (for protein) and 407 nm (for ADMQ) (PTI International Class I Nano LED) as a light source with a TBX-04 detector (all IBH, U.K). The decays were untangled on Felix GX 4.1.2 software. The mean fluorescence lifetimes for biexponential decays were calculated from the decay times (τi) and the relative contribution of components (αi). The qualities of the fits were judged by reduced χ2 criterion and the randomness of the fitted function of the raw data.
Site marker competitive experiments. Site specific marker displacement experiment were performed to identify binding location between ADMQ and BSA in the presence of two site markers (warfarin and ibuprofen) using the fluorescence titration methods. Equimolar concentrations of BSA and warfarin/ibuprofen were used and ADMQ was then gradually added. Fluorescence spectra were recorded at 298 K with an excitation wavelength of 280 nm with emission range of 300–700 nm.
Electrophoresis of BSA–ADMQ. SDS-PAGE in 10% polyacrylamide gel has been performed under reducing conditions. The glass plate sandwich of the electrophoresis apparatus are assembled according to the featured instructions (Biorad) using two clean glass plates. The resolving gel solution is mixed thoroughly and pipetted carefully in the glass plate sandwich leaving a considerable space above the gel for stacking gel. Water saturated isopropanol is layered on top of the polymerizing solution. Next the stacking gel mixture is prepared and pipetted over the top. A 0.75 mm Teflon comb is inserted into the gel solution in between the two glass plates and the gel is allowed to polymerize. The protein samples are prepared and mixed with the previously prepared sample buffer in 1:1 dilution sample buffer in 1:1 dilution. The system is covered and connected to power supply at 15 mA constant current until the bromophenol blue (BPB) tracking dye reaches the bottom of the separating gel. Protein load in gel slots was 5 μg, and gel was stained with Coomassie brilliant blue R. Broad range molecular weight markers was used that included phosphorylase b (97.4 kDa), BSA (66 kDa), ovalbumin (43 kDa), carbonic anhydrase (31 kDa), soyabean trypsin inhibitor (20.1 kDa), and lysozyme (14.3 kDa). Untreated BSA was included as control. BSA and ADMQ at increasing concentrations was mixed and incubated for 12 h before loading onto the gel.
In silico studies. The computational studies are carried out using Linux centos 6 operating system, 64 bit Intel® core™ i5-2500 CPU @ 3.30 GHz, 4 GB RAM using Schrödinger suite 2014 by maestro 9.9 (Schrödinger 2014: Maestro, version 9.9, Schrödinger, LLC, NY, 2014).
Ligand and protein preparation. The newly synthesized ligand ADMQ was sketched in Maestro 9.9 (Maestro, version 9.9, Schrodinger, LLC, New York, NY, 2014), and ligand preparation was carried out with LigPrep module (Schrödinger 2014: LigPrep, version 3.1, Schrödinger, LLC, NY, 2014) using OPLS-2005 force field to rectify the molecular geometries, ionize at biological pH 7.4 to retain specific chirality and to get least energy conformations required for docking.The protein BSA used in the study is retrieved from the protein data bank (PDB), identifier 3V03 which is first prepared by using protein preparation wizard (Schrödinger Release 2014-3: Schrödinger Suite 2014-3 Protein Preparation Wizard; Epik version 2.9, Schrödinger, LLC, NY, 2014; Impact version 6.4, Schrödinger, LLC, NY, 2014; Prime version 3.7, Schrödinger, LLC, NY, 2014.) where it is preprocessed to add hydrogen, filling in missing atoms and loops using prime followed by optimization of the H-bond especially for Asp, Glu, His, hydroxyl containing residues. This model for BSA was hence reliable and most likely resembles to the native three-dimensional structure. At last the complete protein is minimized to the least energy state required for docking by using convergence by RMSD of 0.3 Å with OPLS (optimized potential for liquid simulations) 2005 force field to rectify the molecular geometries and to retain specific chirality.
Identification of active site of bovine serum albumin. The fully prepared protein is investigated for the possible binding sites by identifying druggable active sites using site map which require at least 15 site points per reported site with options to report up to five sites using more restrictive definition of hydrophobicity with setting for standard grid with crop sites at 4 Å from nearest site point. Docking is adopted to determine at first level of approximation a list of possible binding sites on BSA and to obtain the binding energy of the protein–probe complex. This energy based scoring function includes terms accounting for short range van der Waals, electrostatic interactions, hydrogen bonding and penalty scoring function. From the docking calculation, the conformer of the ligand which found to be best complementarity and least penalty scores with minimum binding energy against the receptor is picked up for further analysis.
Molecular docking and binding free energy study. ADMQ is docked at the sites identified by sitemap using Glide ligand docking [Small-Molecule Drug Discovery Suite 2014-3: Glide, version 6.4, Schrödinger, LLC, NY, 2014] using XP mode with flexible ligand sampling where options are with sample ring conformation and nitrogen inversion adding Epik state penalty. Ligand to be docked have a receptor grid scaling of van der Waals radii 0.8 and partial charge cut off 0.15 whose pose viewer files containing the best pose of each ligand in the particular site is generated that poses the more nonbonding interactions which are further analyzed. The pose viewer files generated by docking were used for calculating the binding free energy using Prime MM-GBSA [Prime, version 3.6, Schrödinger, LLC, NY, 2014] with setting of solvation model VSGB with force field OPLS 2005 and the sampling methods were set to do minimization of side chains. In addition to the regular XP docking, we also performed flexible docking using Induced Fit Docking (IFD) (Induced Fit Docking protocol 2014-2, Glide version 6.1, Prime version 3.4, Schrödinger, LLC, NY, 2014) which considers both ligand and receptor flexibility where ligands are docked to the protein using soften potential docking in Glide program and the resulting top poses were used to sample the protein plasticity using the prime program in Schrodinger suite.
Molecular dynamics simulation. The molecular dynamics simulation performed in this study using Desmond (Desmond Molecular Dynamics System, Version 2.2, D.E. Shaw Research, NY, 2009. Maestro-Desmond Interoperability Tools, Version 2.2, Schrodinger, NY, 2009) involves three steps which are system building, minimization followed by NPT ensemble molecular dynamics. The TIP3P solvation model was set with orthorhombic box shape where box volume is calculated and minimized to least possible number. The ions were neutralized by adding Na+ salt with force field OPLS 2005. The solvated ligand receptor complex were further minimized with maximum iterations of 2000 convergence threshold is 1.0 kcal mol−1 Å−1.The minimized solvated ligand receptor complex is processed for molecular dynamics where simulation setting was with simulation period of 10 ns with a recording interval of 1.2 ps with trajectory of 4.8. NPT ensemble method with thermostat Nose–Hoover chain by relaxation time of 1.0 ps with temperature 300 K and pressure 1.01325 bar with facility to relax model before simulation with RESPA integrator by time step bonded 2.0 fs. Coulombic short range method cutoff was with 9 A and long range method of smooth particle mesh Ewald with Ewald tolerance of 10−9 was used in the study.
Conclusions
The present comprehensive study of a quinoline appended chalcone derivative ADMQ indicates that it could induce a lowering of blood pressure via significant inhibition of ACE in a dose dependent manner. ADMQ undergoes strong interaction with ACE both in absence and presence of carrier protein. ADMQ is surrounded by hydrophobic residues inside the pocket of ACE, β-hydroxy group of ADMQ forms H-bonded with Gln 281 and a bridged water H-bond is formed with Tyr 520 where as the carbonyl oxygen is H-bonded with Lys 511 and Ala 354 along with a bridged water H-bond with Phe 512 and Tyr 146. The local vicissitudes along the protein chain is identified by the protein root mean square fluctuation. Analogous ACE inhibitory mechanistic profile of ADMQ with standard drug captopril is recognized in terms of ligand interaction pattern, changes in secondary structural elements and protein RMSF value. The intrinsic fluorescence quenching of BSA signifies strong binding interaction of ADMQ with the same. This phenomenon is recognized as ADMQ–BSA ground state complex formation at physiological pH 7.4 contrary to the time resolved and FRET measurements which designates that both static and energy transfer mechanism co-exists. The ADMQ–BSA binding process is spontaneous in nature and the electrostatic as well as hydrophobic interaction paved way in stabilizing the complex. Higher motional restriction on the fluorophore in proteinaceous microenvironment is manifested by larger fluorescence anisotropy value of 0.206. The probe binds in the warfarin binding site of BSA (BS I) and it lies in close proximity to the Trp 213 residue in the hydrophobic pocket of subdomain II A. The Forster's distance between Trp 213 and ADMQ is found to be 2.45 nm which is further substantiated from induced fit docking studies. The minor decrease of α-helical content obtained from CD spectral measurement and 1–3 Å change in protein RMSD during MD simulation clearly indicate that the polypeptide chain is partially destabilized due to the above site specific accommodation and it may not affect the physiological function of the carrier protein during transportation of this potential antihypertensive molecule. A slight diminution in the ACE inhibitory profile is observed in presence of BSA, however the model carrier protein shows lesser binding towards ADMQ in presence of ACE, which may be beneficial for its delivery to the target enzyme. The present study envisages the usefulness of a heterocyclic chalcone derivative as a therapeutic target in controlling hypertension. This comprehensive ‘in vitro’ and ‘in silico’ research described may open up new avenues for chalcone based drug designing in academic as well as in industry.
Acknowledgements
Financial support from CSIR scheme no. 37 (1493)11/EMR-II is gratefully acknowledged. RJ thanks CSIR, New Delhi for providing financial support [09/1092(0001)/2012-EMR-I]. SKG gives thanks to Prof. S. C. Bhattacharya, Dept of Chemistry, Jadavpur University for giving opportunity of Circular Dichroism (CD) measurement. We would also like to thank respected anonymous reviewers for their critical comments and suggestions.
References
- T. Enoki, H. Ohnogi, K. Nagamine, Y. Kudo, K. Sugiyama, M. Tanabe, E. Kobayashi, H. Sagawa and I. Kato, J. Agric. Food Chem., 2007, 55, 6013–6017 CrossRef CAS PubMed.
- A. Boumendjel, J. Boccard, P.-A. Carrupt, E. Nicolle, M. Blanc, A. Geze, L. Choisnard, D. Wouessidjewe, E.-L. Matera and C. Dumontet, J. Med. Chem., 2008, 51, 2307–2310 CrossRef CAS PubMed.
- T. Akihisa, H. Tokuda, D. Hasegawa, M. Ukiya, Y. Kimura, F. Enjo, T. Suzuki and H. Nishino, J. Nat. Prod., 2006, 69, 38–42 CrossRef CAS PubMed.
- N. Tadigoppula, V. Korthikunta, S. Gupta, P. Kancharla, T. Khaliq, A. Soni, R. K. Srivastava, K. Srivastava, S. K. Puri, K. S. R. Raju, Wahajuddin, P. S. Sijwali, V. Kumar and I. S. Mohammad, J. Med. Chem., 2013, 56, 31–45 CrossRef CAS PubMed.
- J.-H. Kang, J. McRoberts, F. Shi, J. E. Moreno, A. D. Jones and G. A. Howe, Plant Physiol., 2014, 164, 1161–1174 CrossRef CAS PubMed.
- G. G. Duthie, S. J. Duthie and J. A. Kyle, Nutr. Res. Rev., 2000, 13, 79–106 CrossRef CAS PubMed.
- M. G. Hertog, E. J. Feskens, P. C. Hollman, M. B. Katan and D. Kromhout, Lancet, 1993, 342, 1007–1011 CrossRef CAS.
- J. Han, D. Wang, B. Yu, Y. Wang, H. Ren, B. Zhang, Y. Wang and Q. Zheng, Oxid. Med. Cell. Longevity, 2014, 2014, 1–11 CrossRef PubMed.
- G. Avila-Villarreal, O. Hernandéz-Abreu, F. Escalante-Erosa, L. Peña-Rodríguez and S. Estrada-Soto, Planta Med., 2012, 78–PD119 Search PubMed.
- S. N. A. Bukhari, A. M. Butt, M. W. B. Amjad, W. Ahmad, V. H. Shah and A. R. Trivedi, Pak. J. Biol. Sci., 2013, 16, 1368–1372 CrossRef CAS.
- H. U. H. Kinugasa, H. Mizuta and M. Tsukamoto and H. Uno, US Pat.,382 8030 A, 1974.
- S. K. Pal, D. Mandal, D. Sukul, S. Sen and K. Bhattacharyya, J. Phys. Chem. B, 2001, 105, 1438–1441 CrossRef CAS.
- R. Chowdhury, S. Chattoraj, S. Sen Mojumdar and K. Bhattacharyya, Phys. Chem. Chem. Phys., 2013, 15, 16286–16293 RSC.
- U. Mandal, S. Ghosh, G. Mitra, A. Adhikari, S. Dey and K. Bhattacharyya, Chem.–Asian J., 2008, 3, 1430–1434 CrossRef CAS PubMed.
- V. K. Jimsheena and L. R. Gowda, Anal. Chem., 2009, 81, 9388–9394 CrossRef CAS PubMed.
- X. Gao, Am. J. Anal. Chem., 2011, 02, 250–257 CrossRef CAS.
- A. Chakrabarty, A. Mallick, B. Haldar, P. Das and N. Chattopadhyay, Biomacromolecules, 2007, 8, 920–927 CrossRef CAS PubMed.
- T. Peters, Adv. Protein Chem., 1985, 37, 161–245 CrossRef CAS.
- B. X. Huang, H.-Y. Kim and C. Dass, J. Am. Soc. Mass Spectrom., 2004, 15, 1237–1247 CrossRef CAS PubMed.
- V. Lhiaubet-Vallet, Z. Sarabia, F. Boscá and M. A. Miranda, J. Am. Chem. Soc., 2004, 126, 9538–9539 CrossRef CAS PubMed.
- M. C. Jiménez, M. A. Miranda and I. Vayá, J. Am. Chem. Soc., 2005, 127, 10134–10135 CrossRef PubMed.
- P. Bolel, N. Mahapatra and M. Halder, J. Agric. Food Chem., 2012, 60, 3727–3734 CrossRef CAS PubMed.
- S. Datta and M. Halder, J. Phys. Chem. B, 2014, 118, 6071–6085 CrossRef CAS PubMed.
- Z. Liu, X. Zheng, X. Yang, E. Wang and J. Wang, Biophys. J., 2009, 96, 3917–3925 CrossRef CAS PubMed.
- I. B. Squire, K. P. J. O'Kane, N. Anderson and J. L. Reid, Hypertension, 2000, 36, 132–136 CrossRef CAS.
- R. Natesh, S. L. U. Schwager, H. R. Evans, E. D. Sturrock and K. R. Acharya, Biochemistry, 2004, 43, 8718–8724 CrossRef CAS PubMed.
- Q. Wang, L. Qiu, X. Chen, K.-K. Song, Y. Shi and Q.-X. Chen, Bioorg. Med. Chem., 2007, 15, 1568–1571 CrossRef CAS PubMed.
- Y.-L. Xue, T. Miyakawa, Y. Hayashi, K. Okamoto, F. Hu, N. Mitani, K. Furihata, Y. Sawano and M. Tanokura, J. Agric. Food Chem., 2011, 59, 6011–6017 CrossRef CAS PubMed.
- H. Kumar, A. Chattopadhyay, R. Prasath, V. Devaraji, R. Joshi, P. Bhavana, P. Saini and S. K. Ghosh, J. Phys. Chem. B, 2014, 118, 7257–7266 CrossRef CAS PubMed.
- A. Divsalar, M. J. Bagheri, A. A. Saboury, H. Mansoori-Torshizi and M. Amani, J. Phys. Chem. B, 2009, 113, 14035–14042 CrossRef CAS PubMed.
- S. K. Ghosh, S. U. Hossain, S. Bhattacharya and S. C. Bhattacharya, J. Photochem. Photobiol., B, 2005, 81, 121–128 CrossRef CAS PubMed.
- B. J. Bhuyan and G. Mugesh, Org. Biomol. Chem., 2011, 9, 1356–1365 CAS.
- H. Kumar, V. Devaraji, R. Prasath, M. Jadhao, R. Joshi, P. Bhavana and S. K. Ghosh, Spectrochim. Acta, Part A, 2015, 151, 605–615 CrossRef CAS PubMed.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS PubMed.
- D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley and W. Sherman, J. Chem. Theory Comput., 2010, 6, 1509–1519 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, U.S.A., 3rd edn, 2006, ch 8, pp. 278–284 Search PubMed.
- B. K. Paul, A. Samanta and N. Guchhait, J. Phys. Chem. B, 2010, 114, 6183–6196 CrossRef CAS PubMed.
- Y.-J. Hu, Y. Ou-Yang, C.-M. Dai, Y. Liu and X.-H. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef CAS PubMed.
- X. Zhao, R. Liu, Z. Chi, Y. Teng and P. Qin, J. Phys. Chem. B, 2010, 114, 5625–5631 CrossRef CAS PubMed.
- Y. Shu, M. Liu, S. Chen, X. Chen and J. Wang, J. Phys. Chem. B, 2011, 115, 12306–12314 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS.
- A. Singha Roy, D. R. Tripathy, A. K. Ghosh and S. Dasgupta, J. Lumin., 2012, 132, 2943–2951 CrossRef CAS PubMed.
- P. Das, A. Chakrabarty, B. Haldar, A. Mallick and N. Chattopadhyay, J. Phys. Chem. B, 2007, 111, 7401–7408 CrossRef CAS PubMed.
- I. M. Kuznetsova, A. I. Sulatskaya, O. I. Povarova and K. K. Turoverov, PLoS One, 2012, 7, e40845 CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1975, 11, 824–832 CAS.
- K. Takehara, K. Yuki, M. Shirasawa, S. Yamasaki and S. Yamada, Anal. Sci., 2009, 25, 115–120 CrossRef CAS.
- Y. Ni, S. Su and S. Kokot, Anal. Chim. Acta, 2006, 580, 206–215 CrossRef CAS PubMed.
- D. P. Rotella and D. J. Abraham, Burger’s Medicinal Chemistry, Drug Discovery and Development, Wiley, 7th edn, 2010 Search PubMed.
- S. Ghosh, S. Jana and N. Guchhait, J. Phys. Chem. B, 2012, 116, 1155–1163 CrossRef CAS PubMed.
- I. Russo Krauss, F. Sica, C. A. Mattia and A. Merlino, Int. J. Mol. Sci., 2012, 13, 3782–3800 CrossRef PubMed.
- M. Kontoyianni, L. M. McClellan and G. S. Sokol, J. Med. Chem., 2004, 47, 558–565 CrossRef CAS PubMed.
- S. B. Nabuurs, M. Wagener and J. de Vlieg, J. Med. Chem., 2007, 50, 6507–6518 CrossRef CAS PubMed.
- W. Sherman, T. Day, M. P. Jacobson, R. A. Friesner and R. Farid, J. Med. Chem., 2006, 49, 534–553 CrossRef CAS PubMed.
- C. R. W. Guimarães, J. Chem. Theory Comput., 2011, 7, 2296–2306 CrossRef.
- N. Ibrahim, H. Ibrahim, S. Kim, J.-P. Nallet and F. Nepveu, Biomacromolecules, 2010, 11, 3341–3351 CrossRef CAS PubMed.
- G. Lu, S. Ai and J. Li, Langmuir, 2005, 21, 1679–1682 CrossRef CAS PubMed.
- M. Mathew, S. Sreedhanya, P. Manoj, C. T. Aravindakumar and U. K. Aravind, J. Phys. Chem. B, 2009, 113, 1777–1784 CrossRef PubMed.
- B. Chakraborty, A. S. Roy, S. Dasgupta and S. Basu, J. Phys. Chem. A, 2010, 114, 13313–13325 CrossRef CAS PubMed.
- Z. Chi and R. Liu, Biomacromolecules, 2011, 12, 203–209 CrossRef CAS PubMed.
- F. Samari, B. Hemmateenejad, M. Shamsipur, M. Rashidi and H. Samouei, Inorg. Chem., 2012, 51, 3454–3464 CrossRef CAS PubMed.
- G. Zhang, Y. Wang, H. Zhang, S. Tang and W. Tao, Pestic. Biochem. Physiol., 2007, 87, 23–29 CrossRef CAS PubMed.
- B. Ojha and G. Das, J. Phys. Chem. B, 2010, 114, 3979–3986 CrossRef CAS PubMed.
- A. B. Patel, S. Srivastava and R. S. Phadke, J. Biol. Chem., 1999, 274, 21755–21762 CrossRef CAS PubMed.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- R. Swaminathan, G. Krishnamoorthy and N. Periasamy, Biophys. J., 1994, 67, 2013–2023 CrossRef CAS.
- S. Soares, N. Mateus and V. de Freitas, J. Agric. Food Chem., 2007, 55, 6726–6735 CrossRef CAS PubMed.
- T. K. Mukherjee, P. P. Mishra and A. Datta, Chem. Phys. Lett., 2005, 407, 119–123 CrossRef CAS PubMed.
- P. Banerjee, S. Pramanik, A. Sarkar and S. C. Bhattacharya, J. Phys. Chem. B, 2009, 113, 11429–11436 CrossRef CAS PubMed.
- T. Förster, Fluoreszenz organischer Verbindungen, Vandenhoeck & Ruprecht, 1982 Search PubMed.
- D. Cilli, C. Mirasole, R. Pennisi, V. Pallotta, A. D’Alessandro, A. Antoccia, L. Zolla, P. Ascenzi and A. di Masi, PLoS One, 2014, 9, e114651 Search PubMed.
- N. M. Hooper and A. J. Turner, Biochem. J., 1987, 241, 625–633 CAS.
- Z. Chi, R. Liu, Y. Teng, X. Fang and C. Gao, J. Agric. Food Chem., 2010, 58, 10262–10269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08778c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.