
Mono- and tetra-nuclear copper complexes bearing bis(imino)phenoxide derived ligands: catalytic evaluation for benzene oxidation and ROP of ε-caprolactone†
Xue Wanga,
Ke-Qing Zhaoa,
Mark R. J. Elsegoodb,
Timothy J. Priorc,
Xiaoming Liud,
Li Wud,
Sergio Sanze,
Euan K. Brechine and
Carl Redshaw*ac
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu, 610066, China
bChemistry Department, Loughborough University, Loughborough, Leicestershire LE11 3TU, UK
cDepartment of Chemistry, The University of Hull, Cottingham Rd, Hull, HU6 7RX, UK. E-mail: C.Redshaw@hull.ac.uk
dCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, China
eEaStCHEM School of Chemistry, University of Edinburgh, David Brewster Road, Edinburgh, EH9 3FJ, Scotland
First published on 8th June 2015
Abstract
Complexes of the type [Cu(L)2] (1) and [Cu4L2(μ4-O)(OAc)4] (2) have been obtained from the reaction of the phenoxydiimine 1,3-(2,6-R22C6H3N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2-5-R1C6H2OH-2 (LH) (where R1 = Me, tBu, Cl; R2 = Me, iPr) with copper(II) acetate [Cu(OAc)2]; changing the molar ratio of the reactants affords differing amounts of 1 or 2. Reaction of the parent dialdehyde [1,3-(CHO)2-5-MeC6H2OH-2] with [Cu(OAc)2] in the presence of Et3N afforded, following work-up, a polymeric chain (3) comprising {[Cu2(OAc)4]OAc}n, HNEt3 and MeCN. The crystal structures of 1 (R1 = Me, R2 = iPr 1a; R1 = Cl, R2 = iPr 1b), 2 (R1 = Me, R2 = Me 2a; R1 = Me, R2 = iPr 2b; R1 = tBu, R2 = Me 2c; R1 = Cl, R2 = Me 2d; R1 = Cl, R2 = iPr 2e; R1 = tBu, R2 = iPr 2f) and 3 are reported (synchrotron radiation was necessary for 3). The magnetic properties of the cluster 2b are presented. Complexes of type 2 and 3 were screened for the ring opening polymerization (ROP) of ε-caprolactone, with or without benzyl alcohol present, under a variety of conditions, however only trace polymer was isolated. The electrochemistry of all complexes was also investigated, together with their ability to catalyze benzene oxidation (using hydrogen peroxide); although low conversions were observed, the tetra-nuclear complexes exhibited excellent selectivity.
CH)2-5-R1C6H2OH-2 (LH) (where R1 = Me, tBu, Cl; R2 = Me, iPr) with copper(II) acetate [Cu(OAc)2]; changing the molar ratio of the reactants affords differing amounts of 1 or 2. Reaction of the parent dialdehyde [1,3-(CHO)2-5-MeC6H2OH-2] with [Cu(OAc)2] in the presence of Et3N afforded, following work-up, a polymeric chain (3) comprising {[Cu2(OAc)4]OAc}n, HNEt3 and MeCN. The crystal structures of 1 (R1 = Me, R2 = iPr 1a; R1 = Cl, R2 = iPr 1b), 2 (R1 = Me, R2 = Me 2a; R1 = Me, R2 = iPr 2b; R1 = tBu, R2 = Me 2c; R1 = Cl, R2 = Me 2d; R1 = Cl, R2 = iPr 2e; R1 = tBu, R2 = iPr 2f) and 3 are reported (synchrotron radiation was necessary for 3). The magnetic properties of the cluster 2b are presented. Complexes of type 2 and 3 were screened for the ring opening polymerization (ROP) of ε-caprolactone, with or without benzyl alcohol present, under a variety of conditions, however only trace polymer was isolated. The electrochemistry of all complexes was also investigated, together with their ability to catalyze benzene oxidation (using hydrogen peroxide); although low conversions were observed, the tetra-nuclear complexes exhibited excellent selectivity.
Introduction
In recent years, ligand frameworks that are capable of binding two transition metals in close proximity have attracted interest, due primarily to the possibilities of beneficial cooperative effects.1 In the field of lactone polymerization, the coordination/insertion mechanism has drawn analogies with biological systems and in-particular the mechanism of hydrolysis of some metalloenzymes, where one of the metals present can coordinate water thereby lowering its pKa (enhanced nucleophilicity) and generating a hydroxide species. A second metal can then bind the substrate and make it more susceptible to nucleophilic attack. With this in mind, Hillmyer and Tolman probed the potential cooperative influence of the Zn–O–Zn motif in lactide polymerization.2 We have been interested in the coordination chemistry of acyclic Schiff base ligands and their potential to hold metals in close proximity by utilizing the phenolic group to form an M–O–M linkage.3 Furthermore, Sun et al. reported that cobalt and nickel complexes bearing bis(imino)phenolate type ligands are active for the oligomerization of ethylene.4 We were also attracted to the potential catalytic ability of copper; complexes bearing NNO tridentate Schiff bases that have been shown to act as useful catalysts for the copolymerization of carbon dioxide and cyclohexene oxide.5 In terms of a Cu4O core, early structural examples utilizing bis(amino)alcohols were reported by Krebs et al.,6 whilst Chaudhuri et al. have extended such studies to related ONONO-type ligation and have examined the magnetic susceptibility of the resulting Cu2 and Cu4 complexes.7 More recently bis(imino)phenoxy N2O type ligation has been utilized to isolate complexes that can act as catalysts for the oxidation of cyclohexane and toluene,8 and in catecholase-like activity.9 We also note that Pandey et al. have, by employing β-ketoaminato ligands, isolated both mono and tetranuclear copper complexes, the formation of which was dictated by the use of anhydrous conditions or not.10 Herein, we explore the chemistry of the ligand set 1,3-(2,6-R22C6H3NCH)2-5-R1C6H2OH-2 (LH) (where R1 = Me, tBu, Cl; R2 = Me, iPr) towards [Cu(OAc)2] and have structurally characterized both tetranuclear and mononuclear complexes (see Scheme 1), the yield of each product can be controlled by the reaction stoichiometry. The polymeric product resulting from the interaction of [1,3-(CHO)2-5-MeC6H2OH-2] with [Cu(OAc)2] in the presence of Et3N is also reported. In terms of catalysis, the tetranuclear complexes and the polymeric complex were screened for their ability to ring open polymerize (ROP) ε-caprolactone, but results were disappointing. We have also investigated the electrochemistry of these complexes and their ability to catalyze benzene oxidation.
 | ||
| Scheme 1 Copper complexes prepared herein. | ||
Results and discussion
Synthesis
CH)2-4-MeC6H2OH] (LH) and [Cu(OAc)2] (two equivalents) in refluxing toluene afforded, following work-up, large green blocks as the major product (ca. 90%) and thin yellow plates as the minor product (ca. 10%), both of which proved to be suitable for single crystal X-ray diffraction.The minor yellow complex was shown to be the bis-complex [CuL2] (1a), whilst the major green product was found to be the tetranuclear complex [Cu4L2(μ4-O)(OAc)4] (2b). By varying the reaction stoichiometry (L![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu 1:1 for 1a and b; 1:2 for 2a–f, see Experimental), it proved possible to also isolate the monomeric complexes 1b (for R1 = Cl, R2 = iPr) and tetrametallic type complexes for R1 = Me, R2 = Me 2a; R1 = tBu, R2 = Me 2c; R1 = Cl, R2 = Me 2d, R1 = Cl, R2 = iPr 2e; R1 = tBu, R2 = iPr 2f.
:Cu 1:1 for 1a and b; 1:2 for 2a–f, see Experimental), it proved possible to also isolate the monomeric complexes 1b (for R1 = Cl, R2 = iPr) and tetrametallic type complexes for R1 = Me, R2 = Me 2a; R1 = tBu, R2 = Me 2c; R1 = Cl, R2 = Me 2d, R1 = Cl, R2 = iPr 2e; R1 = tBu, R2 = iPr 2f.
For the mononuclear complexes, mass spectra exhibited peaks for [M + H]+, whilst in the IR spectra bands at 1612/1617 and 1542 (for type 1 complexes) and 1617 and ca. 1550 (for type 2) cm−1 were consistent with the presence of the imine CN linkage. In the IR spectra of the ‘free ligands’, two strong absorptions are observed in the region 1580–1630 cm−1 for the imine stretching mode. There is thus a shift to lower frequency upon coordination. In the case of the phenolic C–O band, there is a shift to higher frequency upon coordination (1327–1347 cm−1 in the complexes versus 1254–1263 cm−1 in the ‘free ligands’) (Fig. 1).
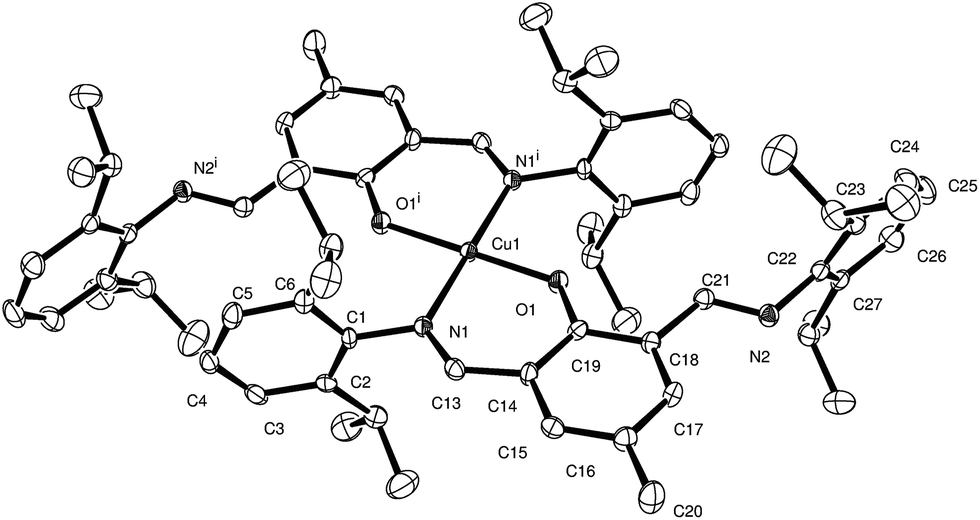 | ||
| Fig. 1 ORTEP representation of the coordination about Cu2+ in 1a, with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–O1 1.917(2), Cu1–N1 1.971(2), N1–C13 1.293(4), N2–C22 1.442(4); O1–Cu1–N2 91.73(9). | ||
The IR spectra of the tetranuclear complexes exhibited a number of weak to medium vC–H bands in the 2860–3020 cm−1 region, together with a weak band at ca. 566 cm−1, which is known to be associated with the T2 mode of the Cu4O.11 In their mass spectra(electrospray), peaks were observed for the fragments resulting from loss of either one or two OAc groups, namely [Cu4(OAc)3(L)2(μ4-O)]+ or [Cu4(OAc)2(L)2(μ4-O)]2+. ESR spectra, recorded at ambient temperature and 103 K, exhibited features similar to that reported by Jian et al. for the complex [Cu4OCl6(C14H12N2)4] (g⊥ = 2.107, g‖ = 2.210);12 g⊥ values herein were found at about g⊥ = 2.42 (see ESI, Fig. S1–S3†).
General. There are two basic modes of coordination of the ligand observed: (1) bidentate coordination by the ligand through phenoxide and nitrogen; (2) bis-bidentate coordination through μ-phenoxide and both imine nitrogen atoms to two copper ions. Bidentate coordination leads to discrete monomeric complexes that feature centrosymmetric binding of the copper ion in a square planar geometry. The bis-bidentate arrangement is found in tetrahedral Cu4O clusters that contain a central oxoanion. A third structure type has been observed in the case of [Et3NH][Cu2(OAc)4](OAc)]·MeCN and this does not contain the Schiff base ligand. [Cu2(OAc)4] paddlewheels are linked by bridging bidentate acetate into a 1-D chain (Fig. 2).
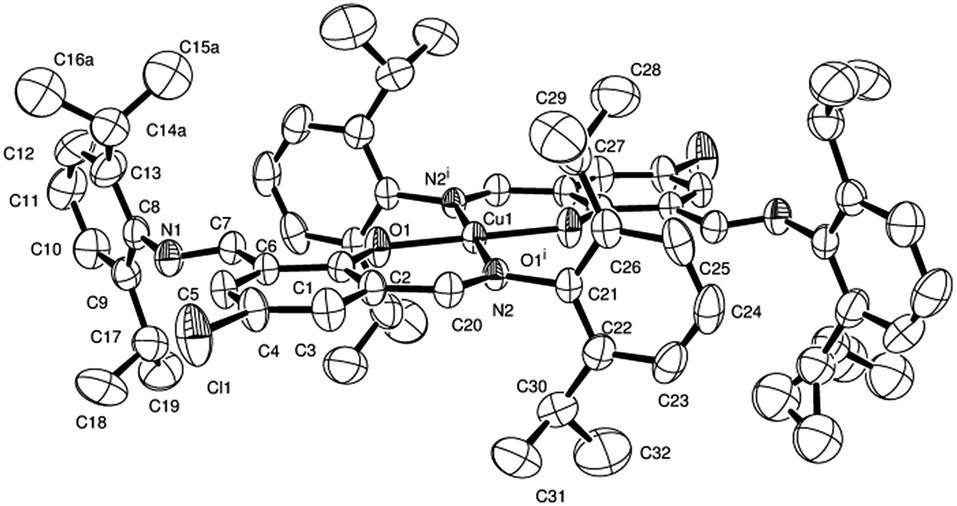 | ||
| Fig. 2 ORTEP representation of the coordination about Cu2+ in 1b, with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–O1 1.9129(15), Cu1–N2 1.9841(17), N1–C7 1.260(3), N1–C8 1.423(3); N2–C20 1.282(3), N2–C21 1.449(3), O1–Cu1–N2 91.36(7). | ||
Monomeric complexes. The compounds 1a and 1b display very similar ligands, differing in the replacement of a methyl group by chloride. As has been noted before, the structural demand of a methyl group and chloride are roughly similar and these two compounds are isomorphous, crystallising in the space group P21/c with similar unit cell parameters. The complex is centrosymmetric with the 4-coordinate square planar Cu2+ ion residing on the inversion centre. For 1a (collected at 160 K) the Cu–O and Cu–N distances are 1.917(2) and 1.971(2) Å and the N–Cu–O bite angle is 91.73(9)°. For 1b (collected at 293 K), the analogous values are 1.9129(15) Å, 1.9841(17) Å, and 91.36(7)°. These geometrical parameters are similar to those reported for related [Cu(N–O)2] type systems; Cu–O 1.889(4) [1.880(5)]*–1.938(5) Å and Cu–N 1.989(7) [1.901(7)]*–2.021(5) Å.13 There are no intermolecular contacts of note for either structure. Crystal data for these are contained in Table 1.
| Identification code | 1a | 1b | 3 |
|---|---|---|---|
| Empirical formula | C66H82CuN4O2 | C64H76Cl2CuN4O2 | C18H34Cu2N2O10 |
| Formula weight | 1026.89 | 1067.72 | 565.55 |
| Temperature | 160(2) K | 293(2) K | 293(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n |
| Unit cell dimensions | a = 9.8597(6) Å α = 90° | a = 10.0099(2) Å α = 90° | a = 12.2105(4) Å α = 90° |
| b = 12.1852(8) Å β = 97.5566(13)° | b = 12.1157(2) Å β = 97.487(2)° | b = 11.5458(4) Å β = 101.407(3)° | |
| c = 24.1112(16) Å γ = 90° | c = 24.5154(5) Å γ = 90° | c = 17.7582(5) Å γ = 90° | |
| Volume | 2871.6(3) Å3 | 2947.80(10) Å3 | 2454.10(14) Å3 |
| Z | 2 | 2 | 4 |
| Density (calculated) | 1.188 Mg m−3 | 1.203 Mg m−3 | 1.531 Mg m−3 |
| Absorption coefficient | 0.427 mm−1 | 0.506 mm−1 | 1.785 mm−1 |
| F(000) | 1102 | 1134 | 1176 |
| Crystal size | 0.64 × 0.05 × 0.02 mm3 | 0.25 × 0.25 × 0.20 mm3 | 0.25 × 0.20 × 0.15 mm3 |
| Theta range for data collection | 1.704 to 25.998° | 2.862 to 28.938° | 2.931 to 28.942° |
| Index ranges | −11 ≤ h ≤ 12, −15 ≤ k ≤ 14, −29 ≤ l ≤ 28 | −13 ≤ h ≤ 12, −15 ≤ k ≤ 13, −33 ≤ l ≤ 20 | −12 ≤ h ≤ 15, −15 ≤ k ≤ 9, −21 ≤ l ≤ 24 |
| Reflections collected | 16148 |
14090 |
11719 |
| Independent reflections | 5606 [R(int) = 0.0835] | 6775 [R(int) = 0.0246] | 5625 [R(int) = 0.0257] |
| Completeness (to 2θ = 25.242°) | 99.6% | 99.8% | 99.8% |
| Absorption correction | Empirical | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. And min. Transmission | 0.992 and 0.772 | 1.000 and 0.958 | 1.000 and 0.835 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 5606/0/340 | 6775/7/331 | 5625/1/301 |
| Goodness-of-fit on F2 | 1.000 | 1.024 | 1.038 |
| Final R indices [I > 2σ(I)] | R1 = 0.0528, wR2 = 0.1035 | R1 = 0.0502, wR2 = 0.1125 | R1 = 0.0359, wR2 = 0.0771 |
| R Indices (all data) | R1 = 0.1074, wR2 = 0.1247 | R1 = 0.0801, wR2 = 0.1270 | R1 = 0.0517, wR2 = 0.0853 |
| Largest diff. Peak and hole | 0.465 and −0.581 e.Å−3 | 0.348 and −0.306 e.Å−3 | 0.374 and −0.499 e.Å−3 |
| Identification code | 2a | 2b | 2c | 2c·MeCN |
|---|---|---|---|---|
| Empirical formula | C66H86Cu4N4O15 | C76H94Cu4N5O11 | C70.67H88Cu4N4O14.33 | C72H86Cu4N8O11 |
| Formula weight | 1429.54 | 1507.72 | 1477.00 | 1493.64 |
| Temperature | 293(2) K | 293(2) K | 293(2) K | 293(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Monoclinic |
| Space group | C2/c | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
| Unit cell dimensions | a = 24.5344(10) Å | a = 20.028(11) Å | a = 14.1438(9) Å | a = 23.6777(5) Å |
| b = 13.0252(4) Å | b = 15.1113(5) Å | b = 14.8853(5) Å | b = 12.2318(3) Å | |
| c = 22.7527(8) Å | c = 25.249(4) Å | c = 20.3062(5) Å | c = 27.2068(6) Å | |
| α = 90° | α = 90° | α = 82.021(2)° | α = 90° | |
| β = 104.984(4)° | β = 98.68(4)° | β = 88.299(3)° | β = 113.205(2)° | |
| γ = 90° | γ = 90° | γ = 64.613(5)° | γ = 90° | |
| Volume | 7023.7(5) Å3 | 7554(5) Å3 | 3822.8(3) Å3 | 7242.2(3) Å3 |
| Z | 4 | 4 | 2 | 4 |
| Density (calculated) | 1.352 Mg m−3 | 1.326 Mg m−3 | 1.284 Mg m−3 | 1.370 Mg m−3 |
| Absorption coefficient | 1.259 mm−1 | 1.171 mm−1 | 1.158 mm−1 | 1.222 mm−1 |
| F(000) | 2984 | 3156 | 1541.3 | 3112 |
| Crystal size | 0.25 × 0.20 × 0.20 mm3 | 0.40 × 0.35 × 0.20 mm3 | 0.35 × 0.25 × 0.25 mm3 | 0.35 × 0.25 × 0.20 mm3 |
| Theta range for data collection | 2.953 to 28.956° | 2.956 to 28.991° | 2.923 to 29.028° | 2.854 to 29.127° |
| Index ranges | −19 ≤ h ≤ 33, −10 ≤ k ≤ 17, −30 ≤ l ≤ 30 | −25 ≤ h ≤ 27, −19 ≤ k ≤ 8, −32 ≤ l ≤ 24 | −18 ≤ h ≤ 19, −20 ≤ k ≤ 20, −27 ≤ l ≤ 26 | −23 ≤ h ≤ 32, −15 ≤ k ≤ 16, −36 ≤ l ≤ 29 |
| Reflections collected | 16990 |
43429 |
33076 |
41758 |
| Independent reflections | 8067 [R(int) = 0.0270] | 17557 [R(int) = 0.0311] |
17406 [R(int) = 0.0463] |
16766 [R(int) = 0.0283] |
| Completeness (to 2θ = 25.242°) | 99.9% | 99.8% | 99.8% | 99.6% |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max. And min. Transmission | 1.000 and 0.787 | 1.000 and 0.840 | 1.000 and 0.843 | 1.000 and 0.881 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 8067/6/386 | 17557/4/859 |
17406/12/838 |
16766/10/880 |
| Goodness-of-fit on F2 | 1.022 | 1.033 | 1.072 | 1.037 |
| Final R indices [I > 2σ(I)] | R1 = 0.0547, wR2 = 0.1321 | R1 = 0.0426, wR2 = 0.0935 | R1 = 0.0736, wR2 = 0.1908 | R1 = 0.0388, wR2 = 0.0837 |
| R Indices (all data) | R1 = 0.0898, wR2 = 0.1537 | R1 = 0.0727, wR2 = 0.1075 | R1 = 0.1248, wR2 = 0.2288 | R1 = 0.0547, wR2 = 0.0914 |
| Largest diff. Peak and hole | 0.849 and −0.658 e.Å−3 | 0.468 and −0.381 e.Å−3 | 1.099 and −0.605 e.Å−3 | 0.728 and −0.463 e.Å−3 |
| Identification code | 2d | 2e | 2f |
|---|---|---|---|
| Empirical formula | C178H183Cl6Cu12N17O33 | C72H88Cl2Cu4N4O11 | C350H183Cu16N31O44 |
| Formula weight | 4063.58 | 1510.52 | 6764.65 |
| Temperature | 293(2) K | 293(2) K | 150(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c | P21 |
| Unit cell dimensions | a = 65.736(3) Å | a = 28.827(2) Å | a = 14.2390(6) Å |
| b = 13.0482(4) Å | b = 28.771(3) Å | b = 51.092(2) Å | |
| c = 22.1089(5) Å | c = 22.942(2) Å | c = 14.3358(5) Å | |
| α = 90° | α = 90° | α = 90° | |
| β = 98.395(2)° | β = 120.445(11)° | β = 118.806(3) | |
| γ = 90° | γ = 90° | γ = 90° | |
| Volume | 18760.4(11) Å3 | 16405(3) Å3 |
9138.7(7) Å3 |
| Z | 4 | 8 | 1 |
| Density (calculated) | 1.439 Mg m−3 | 1.223 Mg m−3 | 1.229 Mg m−3 |
| Absorption coefficient | 1.488 mm−1 | 1.141 mm−1 | 0.976 mm−1 |
| F(000) | 8336 | 6288 | 3536 |
| Crystal size | 0.35 × 0.30 × 0.25 mm3 | 0.20 × 0.15 × 0.15 mm3 | 0.42 × 0.38 × 0.32 mm3 |
| Theta range for data collection | 2.896 to 29.172° | 3.009 to 25.601° | 1.594 to 25.285° |
| Index ranges | −55 ≤ h ≤ 87, −16 ≤ k ≤ 16, −27 ≤ l ≤ 25 | −35 ≤ h ≤ 34, −34 ≤ k ≤ 31, −27 ≤ l ≤ 24 | −17 ≤ h ≤ 17, −58 ≤ k ≤ 61, −15 ≤ l ≤ 17 |
| Reflections collected | 50651 |
35204 |
48175 |
| Independent reflections | 20415 [R(int) = 0.0369] |
15360 [R(int) = 0.0497] |
27844 [R(int) = 0.1154] |
| Completeness (to 2θ = 25.242°) | 98.7% (25.242°) | 99.5% | 98.9% (25.242°) |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents | Empirical |
| Max. And min. Transmission | 1.000 and 0.628 | 1.000 and 0.404 | 1.000 and 0.721 |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 20415/27/1126 |
15360/0/838 |
27844/36/1980 |
| Goodness-of-fit on F2 | 1.050 | 1.038 | 0.874 |
| Final R indices [I > 2σ(I)] | R1 = 0.0497, wR2 = 0.1175 | R1 = 0.0643, wR2 = 0.1577 | R1 = 0.0777, wR2 = 0.1991 |
| R Indices (all data) | R1 = 0.0734, wR2 = 0.1317 | R1 = 0.1204, wR2 = 0.1951 | R1 = 0.1362, wR2 = 0.2448 |
| Largest diff. Peak and hole | 0.918 and −0.632 e.Å−3 | 0.681 and −0.580 e.Å−3 | 0.611 and −1.187e.Å−3 |
| Run | Pre-Cat | T (°C) | CL:BnOH |
Time (h) | Conv.b (%) | Mn,GPCc | Mn,Cald | PDI |
|---|---|---|---|---|---|---|---|---|
| a Runs conducted in toluene using 0.04 mmol of catalyst; CL = ε-caprolactone.b Determined by 1H NMR spectroscopy.c GPC data in THF versus polystyrene standard using a correction factor of 0.56.d Calculated from ([CL]0/[BnOH]0) × conv. (%) × monomer molecular weight. | ||||||||
| 1 | 2a | 40 | 250:1 |
24 | — | — | — | — |
| 2 | 2a | 60 | 250:1 |
24 | — | — | — | — |
| 3 | 2a | 80 | 250:1 |
24 | — | — | — | — |
| 4 | 2a | 100 | 250:1 |
24 | 3 | — | — | — |
| 5 | 2a | 110 | 250:1 |
24 | 3 | 374 | 885 | 1.01 |
| 6 | 2a | 120 | 250:1 |
12 | 10 | — | — | — |
| 7 | 2b | 100 | 800:1 |
12 | 2.2 | 389 | 2006 | 1.01 |
| 8 | 2b | 120 | 250:1 |
24 | 25 | 2936 | 7125 | 1.08 |
| 9 | 2c | 110 | 250:1 |
24 | 22 | 1644 | 6270 | 1.10 |
| 10 | 2c | 120 | 250:1 |
12 | — | — | — | — |
| 11 | 2d | 20 | 250:1 |
24 | 20 | 676 | 5700 | 1.02 |
| 12 | 2e | 110 | 250:1 |
24 | 3.5 | 412 | 998 | 1.01 |
| 13 | 2f | 80 | 250:1 |
24 | — | — | — | — |
| 14 | 2f | 100 | 250:1 |
12 | — | — | — | — |
| 15 | 2f | 120 | 400:1 |
12 | — | — | — | — |
| 16 | 2f | 120 | 600:1 |
12 | — | — | — | — |
| 17 | 2f | 120 | 800:1 |
12 | — | — | — | — |
| 18 | 2f | 120 | 1000:1 |
12 | — | — | — | — |
| 19 | 3 | 25 | 250:1 |
24 | — | — | — | — |
| 20 | 3 | |||||||
Cu4O oxo-clusters. Each of the remaining five ligands (L) forms a similar cluster with formula Cu4OL2(OAc)4 in which each of the copper ions is five coordinated in a square pyramidal geometry. The cluster contains two ligands, each of which binds two copper ions. The ligands are approximately orthogonal and lie on opposite sides of the cluster. Coordination about the copper is completed by bridging bidentate acetate as shown below. Crystal data for these are contained in Table 2. The central μ4-oxo is thought to arise due the presence of adventitious oxygen (Fig. 3).
| Sample | Conversion (%) | Yield (%) | Selectivity (%) | Reduction potential Ep (V) |
|---|---|---|---|---|
| a The substrate (benzene) is 10 mmol; the copper complex (0.02 mmol); conducted in 2.5 ml MeCN. | ||||
| 1a | 31.5 | 12.2 | 39 | −1.32 |
| 2a | 7.2 | 5.5 | 76 | −0.719, −1.019, −1.535 |
| 2b | 7.6 | 5.7 | 75 | |
| 2c | 7.3 | 7.6 | >99 | |
| 2d | 6.9 | 3.9 | 57 | |
| 2e | 7.7 | 8.7 | >99 | |
| 2f | 13.4 | 11.3 | 84 | |
| 3 | 8.5 | 7.0 | 82 | −0.963, −1.598 |
 | ||
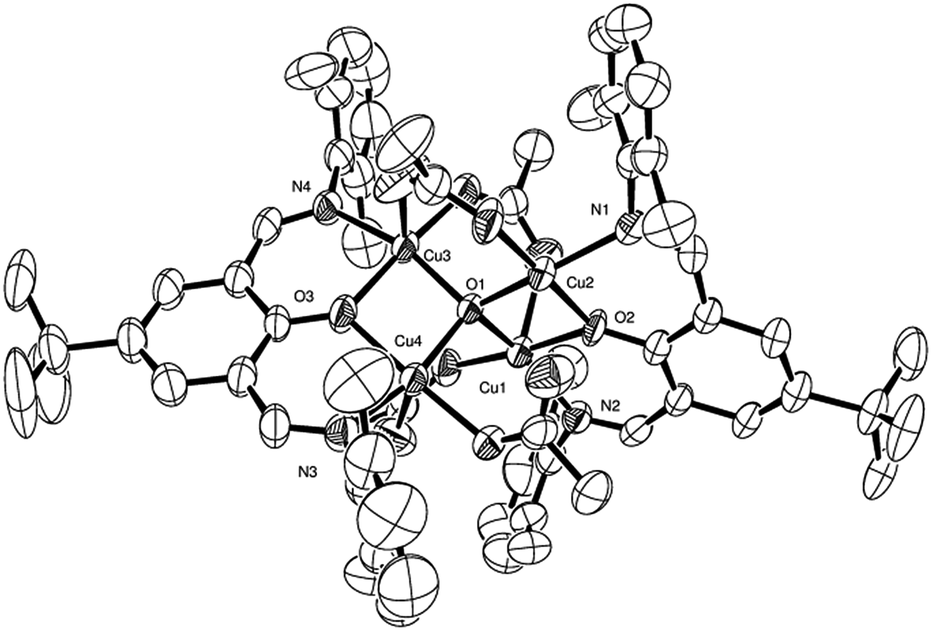
| Fig. 3 ORTEP representation of the cluster in 2a, with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–O1 1.9142(19), Cu1–O2 1.963(2), Cu1–O6 1.930(3), Cu1–N1 1.991(3), Cu1–Cu2 2.9640(6), Cu2–O1 1.9228(16), Cu2–O2 1.988(3), Cu2–O3 1.920(3), Cu2–O5 2.283(3), Cu2–N2 2.001(3), Cu1–O1–Cu2 101.15(2), Cu1–O2–C14 129.3(2), Cu1–O2–Cu2 97.22(11). | ||
The complex 2c is representative of the others. Cu–O1 distances within the oxo-cluster lie in the range 1.910(3) to 1.928(3) Å. Coordination by the ligand through O2 and O3 (i.e. Cu–O2 and Cu–O3) distances lie in the range 1.968(3) to 1.995(3) Å. Copper–nitrogen distances (ligand) lie in the range 1.988(4) to 2.008(4) Å. Each of these copper ions display square-based pyramidal geometry where the apical Cu–O bond is much longer than those bonds in the square plane. For example, for Cu1, Cu–L distances in the plane are 1.928(3), 1.922(4), 1.968(3), 1.993(4) Å, but the apical Cu–O distance is 2.308(4) Å. Each of the structures 2a, 2b, 2c, 2c·MeCN, 2d, 2e and 2f display a very similar cluster and orientation of the pair of ligands. Representations of the clusters in 2a, 2c and 2e are shown in Fig. 3–5, respectively.
 | ||
| Fig. 4 ORTEP representation of the cluster in 2c, with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–O1 1.928(3), Cu1–O2 1.968(3), Cu1–O5 2.308(4), Cu1–N2 1.993(4), Cu1–Cu2 2.9907(10), Cu1–O1–Cu2 102.41(14), Cu1–O2–C14 105.96(16), Cu1–O2–Cu2 98.45(13). | ||
The arrangement of these clusters is unremarkable. There are no hydrogen bonds between the clusters in any case. For some examples, solvent molecules are included in the crystal structure. Full details of stoichiometry and crystal data are found in Table 2.
A search of the Cambridge Crystallographic Database (CSD) for the motif 1,3-(C(R)N)2-C6H3O-2 bound to copper found 16 hits for R = Me. Of the non-macrocyclic examples, there was a preference for dinuclear coordination of the bis(imino)phenoxy ligand set as opposed to the mononuclear coordination observed herein for 1a and 1b; representative examples are given in ref. 14. We note that 1:1 complexes have been isolated from reactions involving CuCl2·H2O and 2,6-bis(imino)phenols.15 In the case of R = H, there were far more hits (356), of which approximately half (48%) were macrocyclic whilst, of the remainder, there were 26 hits involving four copper centres bound to a central μ4-oxo group, 23 of which also involved acetate-type bridging.7,8,14c,16
 | ||
| Fig. 5 ORTEP representation of the cluster in 2e, with thermal ellipsoids at 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Cu1–O1 1.914(3), Cu1–O2 1.992(3), Cu(1)–O(4) 2.235(5), Cu(1)–O(8) 1.908(4), Cu1–N1 2.014(4); Cu1–O1–Cu2 103.14(14), Cu1–O2–Cu2 98.23(14). | ||
 | ||
| Fig. 6 ORTEP representation of a portion of one infinite chain running parallel to c in 3, with thermal ellipsoids at 50% probability level. Selected bond lengths (Å) and angles (°): Cu1–O1 1.9832(17), Cu1–O3 1.9648(18), Cu1–O9 2.1166(15); O1–Cu1–O3 88.72(8). | ||
Between the chains lie Et3NH+ cations and acetonitrile. The Et3NH+ cations form a hydrogen bond to acetate through N1. For the hydrogen bond N1–H1⋯O10, N–H distance = 0.91(2) Å, N⋯O distance = 2.806(3) Å, N–H⋯O angle = 172(2)°. Crystal data for 3 are contained in Table 1.
In the IR spectrum of the polymeric chain {[Cu2(OAc)4]OAc}n (3), it was not possible to distinguish between the two different types of bridging acetate groups; only a strong broad carbonyl stretch at 1603 cm−1 together with a stretch at 1422 cm−1 were observable.
 | ||
| Fig. 7 Cyclic voltammograms of complex 1a (3.2 mmol L−1) in 0.1 mol L−1 [NBut4]BF4–CH3CN under Ar atmosphere (298 K, scanning rate = 100 mV s−1, red dot-line: reduction process; blue dot- and red solid-lines: oxidation). | ||
 | ||
| Fig. 8 Cyclic voltammograms of complex 2a (3.2 mmol L−1) in 0.1 mol L−1 [NtBu4]BF4–CH3CN under Ar atmosphere (298 K, scanning rate = 100 mV s−1, red solid-, blue dash- and purple dot-lines: reduction process; turquoise dot-dash-line: oxidation). | ||
 | ||
| Fig. 9 Cyclic voltammograms of complex 3 (3.2 mmol L−1) in 0.1 mol L−1 [NBut4]BF4–CH3CN under Ar atmosphere (298 K, scanning rate = 100 mV s−1 red dot- and blue solid-lines: reduction process; purple dot-dash-line: anodic region). | ||
The room temperature value of 0.8 cm3 mol−1 K is well below that expected for four non-interacting s = 1/2 ions with g = 2.0 (1.5 cm3 K mol−1). As the temperature is decreased the value of χMT decreases rapidly to a value of 0.1 cm3 K mol−1 at T = 100 K, before decreasing more slowly to a value of zero at the lowest temperature measured. This behaviour is indicative of the presence of strong antiferromagnetic interactions between the Cu(II) ions. The experimental susceptibility data can be fitted to the simple 2J model shown in the inset of Fig. 10 and expressed in isotropic spin-Hamiltonian (1) in which J1 corresponds to the Cu⋯Cu interaction mediated through one oxo and one carboxylate bridge, and J2 that mediated via one oxo and one alkoxo bridge. The best fit afforded J1 = −192 cm−1, J2 = −61 cm−1 with g = 2.2, values consistent with those previously seen for similar species.6b,7,10 This results in a spin singlet ground state (S = 0) with the first excited (S = 1) triplet state some 384 cm−1 higher in energy.
| Ĥex = −2J1(Ŝ1Ŝ3 + Ŝ1Ŝ4 + Ŝ2Ŝ3 + Ŝ2Ŝ4) − 2J2(Ŝ1Ŝ2 + Ŝ3Ŝ4) | (1) |
 | ||
| Fig. 10 Plot of the χMT product versus T for complex 2b in the T = 300–5 K range in an applied field of 0.1 T. The solid red line is a fit of the experimental data to the isotropic model shown schematically in the inset, and spin-Hamiltonian (1). | ||
:2 ratio at 7.35 and 5.10 ppm suggested that there was a benzyl ester cap present.Due to these disappointing results, no further investigation of the potential of these complexes for ring opening polymerization was conducted.
Conclusion
In conclusion, both mononuclear and tetranuclear complexes are accessible form the reaction of 2,6-(2,6-iPr2C6H3NCH)2-4-MeC6H2OH] (LH) and [Cu(OAc)2], the yields of which can be controlled by variation of the reaction stoichiometry. Changing the substituents at the ortho position of the aryl (imino) ring leads to slight variations in the tetrametallic core. The tetrametallic complexes were found to be virtually inactive for the ring opening polymerization of ε-caprolactone in the presence of benzyl alcohol. Magnetic susceptibility studies on the tetrametallic complex with R1 = Me and R2 = iPr indicated the presence of strong antiferromagnetic interactions between the Cu(II) ions. The complexes catalyze the hydroxylation of benzene into phenol by hydrogen peroxide. The yields were not superior to others recently reported, however the tetra-nuclear complexes exhibited excellent (near quantitative) selectivities.
Experimental
General
All manipulations were carried out under an atmosphere of dry nitrogen using conventional Schlenk and cannula techniques or in a conventional nitrogen-filled glove box. Diethyl ether and tetrahydrofuran were refluxed over sodium and benzophenone. Toluene was refluxed over sodium. Dichloromethane and acetonitrile were refluxed over calcium hydride. All solvents were distilled and degassed prior to use. IR spectra (nujol mulls, KBr or NaCl windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer; elemental analyses were performed by the elemental analysis service at Sichuan Normal University. Ligands of type LH were prepared as described in the literature.7 All other chemicals were purchased from commercial sources and were used as received.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH)2-5-MeC6H2O-2]2Cu (1a). 2,6-Diformyl-4-methyl-phenoxy(2,6-diisopropylaniline) (0.49 g, 1.0 mmol) and copper diacetate (0.18 g, 1.0 mmol) in toluene (30 ml) were refluxed under an argon atmosphere for 12 h. The solvent was then removed in vacuo and the residue was extracted into either hot acetonitrile (25 ml) or ethanol (25 ml). Prolonged standing at ambient temperature afforded 1a as green/yellow crystals. Yield: 0.18 g, 35%; elemental analysis calculated for C66H82CuN4O2: C, 77.19; H, 8.05; N, 5.46. Found: C, 76.68; H, 8.15; N, 5.22%. IR (nujol null, KBr): 3059(w), 3005(w), 2959(s), 2926(s), 2867(s), 2357(w), 1923(w), 1617(s), 1601(w), 1587(w), 1542(s), 1443(s), 1381(s), 1364(s), 1342(m), 1326(m), 1294(m), 1258(s), 1228(s), 1180(s), 1165(m), 1108(w), 1097(w), 1043(w), 978(w), 934(w), 820(m), 798(m), 775(s), 761(w), 702(w), 638(w), 561(w), 520(s), 474(w); MS (ESI): m/z 1026 M+.CH)2-5-ClC6H2O-2]2Cu (1b). As for 1a, but using 2,6-diformyl-4-chloro-phenoxy(2,6-diisopropylaniline) (0.51 g, 1.0 mmol) and copper diacetate (0.18 g, 1.0 mmol) affording 1b as green/yellow crystals. Yield: 0.22 g, 41%. elemental analysis calculated for C64H76Cl2CuN4O2: C, 71.99; H, 7.17; N, 5.25. Found: C, 72.16; H, 7.32; N, 5.13%. IR (nujol null, KBr): 3066(s), 2961(s), 2095(s), 2866(m), 2356(w), 1737(w), 1612(s), 1588(w), 1542(s), 1436(s), 1385(m), 1367(s), 1332(s), 1292(w), 1256(m), 1215(s), 1172(s), 1097(m), 1059(w), 1044(w), 1023(s), 932(s), 871(w), 862(w), 797(s), 771(m), 756(s), 728(s), 697(w), 660(w), 638(w), 562(w), 533(m), 510(m), 481(w). MS (ESI): m/z 1068 [M + H]+.CH)2-5-MeC6H2O-2]2(μ4-O)} (2a). A toluene solution (30 ml) of 2,6-diformyl-4-methyl-phenoxy(2,6-dimethylaniline) (0.37 g, 1.0 mmol), copper diacetate (0.37 g, 2.0 mmol) was refluxed under an argon atmosphere for 12 h. The solvent was then removed in vacuo and the residue extracted in hot acetonitrile (30 ml) or ethanol (30 ml). Prolonged standing (2–3 days) afforded 2a as green crystals. Yield: 0.36 g, 57%; elemental analysis calculated for C58H62Cu4N4O11: C, 55.94; H, 5.02; N, 4.50. Found: C, 55.63; H, 5.25; N, 4.31%. IR (nujol null, KBr): 3447(s), 2924(m), 2856(w), 2362(s), 2337(s), 1612(s), 1549(m), 1452(m), 1394(s), 1341(w), 1262(w), 1181(m), 1074(s), 830(w), 769(w), 719(w), 669(w), 566(w), 517(w), 489(w). MS (ESI): m/z 1312 {Cu4(OAc)2[1,3-(2,6-Me2C6H3NCH)2-5-MeC6H2O-2]2(μ4-O)}2+.CH)2-5-MeC6H2O-2]2(μ4-O)} (2b). As for 2a, but using 2,6-diformyl-4-methyl-phenoxy-(2,6-diisopropylaniline) (0.49 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) afforded 2b as green crystals. Yield: 0.44 g, 60%; elemental analysis calculated for C74H94Cu4N4O11: C, 60.47; H, 6.45; N, 3.81. Found: C, 60.53; H, 6.93; N, 4.01%. IR (nujol null, KBr): 3064(w), 2962(s), 2925(s), 2867(s), 2357(w), 1923(w), 1784(w), 1612(s), 1587(s), 1550(s), 1456(s), 1450(m), 1397(s), 1327(s), 1258(m), 1232(w), 1174(s), 1104(m), 1070(s), 1014(m), 932(w), 867(w), 828(w), 800(s), 770(m), 727(m), 665(m), 620(m), 564(m), 528(m), 487(s), 425(w). MS (ESI): m/z 1408 {Cu4(OAc)3[1,3-(2,6-iPr2C6H3NCH)2-5-MeC6H2O-2]2(μ4-O)}+.CH)2-5-tBuC6H2O-2]2(μ4-O)} (2c). As for 2a, but using 2,6-diformyl-4-t-butyl-phenoxy(2,6-dimethylaniline) (0.42 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2c as green crystals. Crystals suitable for single crystal X-ray diffraction can be grown from either methanol or acetonitrile. Yield: 0.32 g, 48%; elemental analysis calculated for C64H74Cu4N4O11: C, 57.82; H, 5.61; N, 4.21. Found: C, 57.71; H, 5.69; N, 4.14%. IR (nujol null, KBr): 3422(s), 3020(w), 2962(s), 2924(s), 2869(w), 2361(w), 1611(s), 1548(m), 1471(m), 1451(m), 1397(s), 1347(m), 1294(w), 1232(m), 1183(s), 1092(w), 1068(s), 1023(w), 923(w), 891(w), 868(w), 841(w), 797(w), 768(s), 722(w), 667(m), 630(m), 566(m), 510(m), 438(m). MS (ESI): m/z 1247 {Cu4(OAc)2[1,3-(2,6-Me2C6H3NCH)2-5-tBuC6H2O-2]2(μ4-O)}2+.CH)2-5-ClC6H2O-2]2(μ4-O)} (2d). As for 2a, but using 2,6-diformyl-4-chloride-phenoxy(2,6-dimethylaniline) (0.39 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2d as green crystals. Yield: 0.37 g, 58%; elemental analysis calculated for C56H56Cl2Cu4N4O11: C, 52.30; H, 4.39; N, 4.36. Found: C, 52.64; H, 4.51; N, 4.53%. IR (nujol null, KBr): 3437(s), 2973(w), 2918(w), 1612(s), 1546(m), 1471(w), 1440(m), 1395(s), 1342(m), 1316(w), 1258(w), 1226(m), 1179(m), 1092(w), 1059(s), 984(w), 924(w), 883(w), 808(m), 766(w), 720(w), 668(m), 627(m), 565(w), 515(w), 438(w). MS (ESI): m/z 1227 {Cu4(OAc)3[1,3-(2,6-Me2C6H3NCH)2-5-ClC6H2O-2]2(μ4-O)}2+.CH)2-5-ClC6H2O-2]2(μ4-O)} (2e). As for 2a, but using 2,6-diformyl-4-chloro-phenoxy(2,6-diisopropylaniline) (0.51 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2e as green crystals. Yield: 0.35 g, 46%; elemental analysis calculated for C72H88Cl2Cu4N4O11: C, 57.25; H, 5.87; N, 3.71. Found: C, 57.36; H, 6.02; N, 3.88%. IR (nujol null, KBr): 3065(w), 2962(s), 2927(s), 2868(s), 2360(w), 1931(w), 1860(w), 1777(w), 1613(s), 1587(s), 1548(s), 1440(s), 1398(s), 1347(s), 1256(w), 1221(w), 1172(s), 1107(m), 1058(s), 990(w), 932(w), 882(w), 863(w), 791(s), 768(w), 724(s), 667(m), 621(s), 563(m). MS (ESI): m/z 991.5 (M+ − L − O).CH)2-5-tBuC6H2O-2]2(μ4-O)} (2f). As for 2a, but using 2,6-diformyl-4-t-butyl-phenoxy(2,6-diisopropylaniline) (0.52 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2f as green crystals. Yield: 0.52 g, 67%; elemental analysis calculated for C80H106Cu4N4O11: C, 61.83; H, 6.88; N, 3.61. Found: C, 61.47; H, 6.96; N, 3.55%. IR (nujol null, KBr): 3447(s), 2962(s), 2869(w), 1612(s), 1553(m), 1458(s), 1397(s), 1360(w), 1331(w), 1261(s), 1175(w), 1097(s), 1068(s), 1023(s), 931(w), 865(w), 804(s), 725(w), 666(w), 623(w), 566(w), 530(w); MS (ESI): m/z 1485 {Cu4(OAc)3[1,3-(2,6-iPr2C6H3NCH)2-5-tBuC6H2O-2]2(μ4-O)}+.
CH)2-5-MeC6H2O-2]2Cu (1a). 2,6-Diformyl-4-methyl-phenoxy(2,6-diisopropylaniline) (0.49 g, 1.0 mmol) and copper diacetate (0.18 g, 1.0 mmol) in toluene (30 ml) were refluxed under an argon atmosphere for 12 h. The solvent was then removed in vacuo and the residue was extracted into either hot acetonitrile (25 ml) or ethanol (25 ml). Prolonged standing at ambient temperature afforded 1a as green/yellow crystals. Yield: 0.18 g, 35%; elemental analysis calculated for C66H82CuN4O2: C, 77.19; H, 8.05; N, 5.46. Found: C, 76.68; H, 8.15; N, 5.22%. IR (nujol null, KBr): 3059(w), 3005(w), 2959(s), 2926(s), 2867(s), 2357(w), 1923(w), 1617(s), 1601(w), 1587(w), 1542(s), 1443(s), 1381(s), 1364(s), 1342(m), 1326(m), 1294(m), 1258(s), 1228(s), 1180(s), 1165(m), 1108(w), 1097(w), 1043(w), 978(w), 934(w), 820(m), 798(m), 775(s), 761(w), 702(w), 638(w), 561(w), 520(s), 474(w); MS (ESI): m/z 1026 M+.CH)2-5-ClC6H2O-2]2Cu (1b). As for 1a, but using 2,6-diformyl-4-chloro-phenoxy(2,6-diisopropylaniline) (0.51 g, 1.0 mmol) and copper diacetate (0.18 g, 1.0 mmol) affording 1b as green/yellow crystals. Yield: 0.22 g, 41%. elemental analysis calculated for C64H76Cl2CuN4O2: C, 71.99; H, 7.17; N, 5.25. Found: C, 72.16; H, 7.32; N, 5.13%. IR (nujol null, KBr): 3066(s), 2961(s), 2095(s), 2866(m), 2356(w), 1737(w), 1612(s), 1588(w), 1542(s), 1436(s), 1385(m), 1367(s), 1332(s), 1292(w), 1256(m), 1215(s), 1172(s), 1097(m), 1059(w), 1044(w), 1023(s), 932(s), 871(w), 862(w), 797(s), 771(m), 756(s), 728(s), 697(w), 660(w), 638(w), 562(w), 533(m), 510(m), 481(w). MS (ESI): m/z 1068 [M + H]+.CH)2-5-MeC6H2O-2]2(μ4-O)} (2a). A toluene solution (30 ml) of 2,6-diformyl-4-methyl-phenoxy(2,6-dimethylaniline) (0.37 g, 1.0 mmol), copper diacetate (0.37 g, 2.0 mmol) was refluxed under an argon atmosphere for 12 h. The solvent was then removed in vacuo and the residue extracted in hot acetonitrile (30 ml) or ethanol (30 ml). Prolonged standing (2–3 days) afforded 2a as green crystals. Yield: 0.36 g, 57%; elemental analysis calculated for C58H62Cu4N4O11: C, 55.94; H, 5.02; N, 4.50. Found: C, 55.63; H, 5.25; N, 4.31%. IR (nujol null, KBr): 3447(s), 2924(m), 2856(w), 2362(s), 2337(s), 1612(s), 1549(m), 1452(m), 1394(s), 1341(w), 1262(w), 1181(m), 1074(s), 830(w), 769(w), 719(w), 669(w), 566(w), 517(w), 489(w). MS (ESI): m/z 1312 {Cu4(OAc)2[1,3-(2,6-Me2C6H3NCH)2-5-MeC6H2O-2]2(μ4-O)}2+.CH)2-5-MeC6H2O-2]2(μ4-O)} (2b). As for 2a, but using 2,6-diformyl-4-methyl-phenoxy-(2,6-diisopropylaniline) (0.49 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) afforded 2b as green crystals. Yield: 0.44 g, 60%; elemental analysis calculated for C74H94Cu4N4O11: C, 60.47; H, 6.45; N, 3.81. Found: C, 60.53; H, 6.93; N, 4.01%. IR (nujol null, KBr): 3064(w), 2962(s), 2925(s), 2867(s), 2357(w), 1923(w), 1784(w), 1612(s), 1587(s), 1550(s), 1456(s), 1450(m), 1397(s), 1327(s), 1258(m), 1232(w), 1174(s), 1104(m), 1070(s), 1014(m), 932(w), 867(w), 828(w), 800(s), 770(m), 727(m), 665(m), 620(m), 564(m), 528(m), 487(s), 425(w). MS (ESI): m/z 1408 {Cu4(OAc)3[1,3-(2,6-iPr2C6H3NCH)2-5-MeC6H2O-2]2(μ4-O)}+.CH)2-5-tBuC6H2O-2]2(μ4-O)} (2c). As for 2a, but using 2,6-diformyl-4-t-butyl-phenoxy(2,6-dimethylaniline) (0.42 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2c as green crystals. Crystals suitable for single crystal X-ray diffraction can be grown from either methanol or acetonitrile. Yield: 0.32 g, 48%; elemental analysis calculated for C64H74Cu4N4O11: C, 57.82; H, 5.61; N, 4.21. Found: C, 57.71; H, 5.69; N, 4.14%. IR (nujol null, KBr): 3422(s), 3020(w), 2962(s), 2924(s), 2869(w), 2361(w), 1611(s), 1548(m), 1471(m), 1451(m), 1397(s), 1347(m), 1294(w), 1232(m), 1183(s), 1092(w), 1068(s), 1023(w), 923(w), 891(w), 868(w), 841(w), 797(w), 768(s), 722(w), 667(m), 630(m), 566(m), 510(m), 438(m). MS (ESI): m/z 1247 {Cu4(OAc)2[1,3-(2,6-Me2C6H3NCH)2-5-tBuC6H2O-2]2(μ4-O)}2+.CH)2-5-ClC6H2O-2]2(μ4-O)} (2d). As for 2a, but using 2,6-diformyl-4-chloride-phenoxy(2,6-dimethylaniline) (0.39 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2d as green crystals. Yield: 0.37 g, 58%; elemental analysis calculated for C56H56Cl2Cu4N4O11: C, 52.30; H, 4.39; N, 4.36. Found: C, 52.64; H, 4.51; N, 4.53%. IR (nujol null, KBr): 3437(s), 2973(w), 2918(w), 1612(s), 1546(m), 1471(w), 1440(m), 1395(s), 1342(m), 1316(w), 1258(w), 1226(m), 1179(m), 1092(w), 1059(s), 984(w), 924(w), 883(w), 808(m), 766(w), 720(w), 668(m), 627(m), 565(w), 515(w), 438(w). MS (ESI): m/z 1227 {Cu4(OAc)3[1,3-(2,6-Me2C6H3NCH)2-5-ClC6H2O-2]2(μ4-O)}2+.CH)2-5-ClC6H2O-2]2(μ4-O)} (2e). As for 2a, but using 2,6-diformyl-4-chloro-phenoxy(2,6-diisopropylaniline) (0.51 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2e as green crystals. Yield: 0.35 g, 46%; elemental analysis calculated for C72H88Cl2Cu4N4O11: C, 57.25; H, 5.87; N, 3.71. Found: C, 57.36; H, 6.02; N, 3.88%. IR (nujol null, KBr): 3065(w), 2962(s), 2927(s), 2868(s), 2360(w), 1931(w), 1860(w), 1777(w), 1613(s), 1587(s), 1548(s), 1440(s), 1398(s), 1347(s), 1256(w), 1221(w), 1172(s), 1107(m), 1058(s), 990(w), 932(w), 882(w), 863(w), 791(s), 768(w), 724(s), 667(m), 621(s), 563(m). MS (ESI): m/z 991.5 (M+ − L − O).CH)2-5-tBuC6H2O-2]2(μ4-O)} (2f). As for 2a, but using 2,6-diformyl-4-t-butyl-phenoxy(2,6-diisopropylaniline) (0.52 g, 1.0 mmol) and copper diacetate (0.37 g, 2.0 mmol) affording 2f as green crystals. Yield: 0.52 g, 67%; elemental analysis calculated for C80H106Cu4N4O11: C, 61.83; H, 6.88; N, 3.61. Found: C, 61.47; H, 6.96; N, 3.55%. IR (nujol null, KBr): 3447(s), 2962(s), 2869(w), 1612(s), 1553(m), 1458(s), 1397(s), 1360(w), 1331(w), 1261(s), 1175(w), 1097(s), 1068(s), 1023(s), 931(w), 865(w), 804(s), 725(w), 666(w), 623(w), 566(w), 530(w); MS (ESI): m/z 1485 {Cu4(OAc)3[1,3-(2,6-iPr2C6H3NCH)2-5-tBuC6H2O-2]2(μ4-O)}+.Electrochemistry
Electrochemistry was performed in a gas-tight three-electrode system in which a vitreous carbon disk (Φ = 1 mm) was used as a working electrode, a carbon strip as counter electrode, and Ag/AgCl (inner reference solution: 0.45 mol L−1 [NtBu4]BF4 + 0.05 mol L−1 [NtBu4]Cl in dichloromethane) against which the potential of ferrocenium/ferrocene couple is 0.55 V in 0.5 mol L−1 [NtBu4]BF4 in CH3CN. Ferrocene was added as an internal standard and all potentials are quoted against ferrocenium/ferrocene couple (Fc+/Fc).Procedure for ROP of ε-caprolactone
Typical polymerization procedures in the presence of one equivalent of benzyl alcohol (Table 3) are as follows. A toluene solution of 2 (0.04 mmol) and benzyl alcohol (0.04 mmol) were added into a Schlenk tube in the glove box at room temperature. The solution was stirred for 2 min, and then ε-caprolactone (10.0 mmol) along with 2 ml toluene was added to the solution. The reaction mixture was then placed into an oil bath pre-heated to the required temperature, and the solution was stirred for the prescribed time. The polymerization mixture was then quenched by the addition of excess glacial acetic acid (0.2 ml) and the resultant solution then poured into methanol (200 ml). The resultant polymer was then dried on filter paper was dried in vacuo.Procedure for oxidation of benzene
Benzene (0.9 ml, 10 mmol), acetonitrile (2.5 ml) and catalytic amount of the copper complex (0.02 mmol) were placed into a reaction vessel equipped with cooling condenser and placed in an oil-bath. The reaction was heated at appropriate temperature for a period of time. When the reaction reached the specified temperature, the appropriate amount of aqueous H2O2 (1.5 ml, 30 wt%) was slowly and carefully added in one-go. When the reaction was stopped and cooled, the volume of the reaction mixture was calibrated to 10 ml with CH3CN in which there was an appropriate amount of toluene as an internal standard. To the calibrated reaction solution was added MgSO4 (3 g) to remove the water in the reaction before being analyzed by gas chromatography. Quantitative analysis of both benzene and phenol was achieved by establishing their calibration curves with two linear equations under optimized conditions, Ab = 0.0053Wb + 0.1266 (R = 0.9986) and Ap = 0.0034Wp − 0.1635 (R = 0.9943) for benzene and phenol, respectively (Fig. S7 and S8, ESI†), where A is the ratio of the peak areas of the analyte (benzene or phenol) and the internal standard toluene, W (mg) is the mass of the analytes, and the subscripts b and p denote benzene and phenol, respectively. The yield of phenol and benzene conversion was calculated as follows: phenol (mmol)/benzene initially used (mmol) × 100% and benzene-reacted (mmol)/benzene initially used (mmol) × 100%, respectively.Crystallography details
For 1a. Single crystal X-ray diffraction data were collected using a Bruker SMART 1K CCD diffractometer using ω scans with narrow frames. Crystals were mounted at the end of a glass fibre under and held at 160 K in an Oxford Cryosystems nitrogen gas Cryostream.
All other samples (except 3). Single crystal X-ray diffraction data were collected at room temperature (293 K) using an Agilent Technologies Xcalibur diffractometer operating with Mo Kα radiation and an Eos CCD detector in a series of ω-scans.20 Data were integrated using standard procedure using CrysAlisPro software (Agilent). Data were corrected for absorption effects using a multi-scan method based on equivalents. In the case of 2f, the crystals appeared to be unstable in the X-ray beam, presumably due to solvent loss. Given this, data for 2f were collected at 150 K, in contrast to the other structures and were coated in a thin film of perfluoropolyether oil. Furthermore, the structure of 2f at room temperature appears different to that at 150 K. At room temperature, the structure is triclinic and centrosymmetric with a single copper cluster in the asymmetric unit. Upon cooling below about 240 K, a larger monoclinic cell of approximately twice the volume emerges. This low temperature form is non-centric and has two unique oxo clusters in the asymmetric unit.
For 3. Data were collected on a Bruker SMART 1K CCD diffractometer at Daresbury SRS station 9.8 (λ = 0.6710 Å).21
Acknowledgements
Sichuan Normal University and the National Natural Science Foundation of China (grants 51443004 and 51273133) are thanked for financial support. The Special Funds for sharing large precision equipment (no. DJ2014-25) at Sichuan Normal University is also thanked. The CCLRC is thanked for the award of beam-time at SRS Daresbury Laboratory (Station 9.8), and Dr Simon J. Teat is thanked for scientific support.Notes and references
- I. Bratko and M. Gómez, Dalton Trans., 2013, 10664 RSC and references therein.
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132 RSC.
- A. Arbaoui, C. Redshaw, N. M. Sanchez-Ballester, M. R. J. Elsegood and D. L. Hughes, Inorg. Chim. Acta, 2011, 365, 96 CrossRef CAS PubMed.
- L. Wang, W.-H. Sun, L. Han, Z. Li, Y. Hu, C. He and C. Yan, J. Organomet. Chem., 2002, 650, 59 CrossRef CAS.
- C.-Y. Tsai, B.-H. Huang, M.-W. Hsiao, C.-C. Lin and B.-T. Ko, Inorg. Chem., 2014, 53, 5109 CrossRef CAS PubMed.
- (a) S. Teipel, K. Griesar, W. Haase and B. Krebs, Inorg. Chem., 1994, 33, 456 CrossRef CAS; (b) J. Reim, K. Griesar, W. Haase and B. Krebs, J. Chem. Soc., Dalton Trans., 1995, 2649 RSC.
- S. Mukherjee, T. Weyhermüller, E. Bothe, K. Wieghardt and P. Chaudhuri, Eur. J. Inorg. Chem., 2003, 863 CrossRef CAS PubMed.
- P. Roy and M. Manassero, Dalton Trans., 2010, 1539 RSC.
- T. Ghosh, J. Adhikary, P. Chakraborty, P. K. Sukul, M. Sekhar Jana, T. K. Mondal, E. Zangrando and D. Das, Dalton Trans., 2014, 841 RSC.
- A. Kumar, R. Pandey, R. K. Gupta, M. Dubey and D. S. Pandey, Dalton Trans., 2014, 13169 RSC.
- (a) J. T. Guy Jr, J. C. Cooper, R. D. Gilardi and J. L. Flippen-Anderson, Inorg. Chem., 1988, 27, 635 CrossRef; (b) A. R. Paital, P. K. Nanda, S. Das, G. Aromi and D. Ray, Inorg. Chem., 2006, 45, 505 CrossRef CAS PubMed.
- F.-F. Jian, P.-S. Zhao, H.-X. Wang and L. D. Wu, Bull. Korean Chem. Soc., 2004, 25, 673 CrossRef CAS.
- (a) P. Mukherjee, M. G. B. Drew and A. Figuerola, Polyhedron, 2008, 27, 3343 CrossRef CAS PubMed; (b) H. Karabiyuk, O. Erdem and M. Aygun, J. Inorg. Organomet. Polym., 2010, 20, 142 CrossRef; (c) L. Mouni, M. Akkurt and S. O. Yildirm, J. Chem. Crystallogr., 2010, 40, 169 CrossRef CAS PubMed; (d) A. Kumar, M. Dubey, R. Pandey, R. K. Gupta, A. Kumar, A. C. Kalita and D. S. Pandey, Inorg. Chem., 2014, 53, 4944 CrossRef CAS PubMed The values in square brackets are for a system exhibiting significant tautomerism.13b.
- (a) N. A. Bailey, D. E. Fenton, J. Lay, P. B. Roberts, J.-M. Latour and D. Limosin, J. Chem. Soc., Dalton Trans., 1986, 2681 RSC; (b) N. E. Borisova, M. D. Reshetova, T. V. Magdesieva, V. N. Khrustalev, G. G. Aleksandrov, M. Kuznetsov, R. S. Skazov, A. V. Dolganov, V. N. Ikorskiy, V. M. Novotortsev, I. L. Eremenko, I. I. Moiseev and Y. A. Ustynyuk, Inorg. Chim. Acta, 2008, 361, 2032 CrossRef CAS PubMed; (c) M.-F. Zaltariov, M. Alexandru, M. Cazacu, S. Shova, G. Novitchi, C. Train, A. Dobrov, M. V. Kirillova, E. C. B. A. Alegria, A. J. L. Pombeiro and V. B. Arion, Eur. J. Inorg. Chem., 2014, 4946 CrossRef CAS PubMed; (d) M. Pait, M. Shatruk, J. Lengyel, S. Gómez-Coca, A. Bauzá, A. Frontera, V. Bertolasi and D. Ray, Dalton Trans., 2015, 44, 9691–9692 RSC.
- L. Han, Y. Cui, Y. Li, W.-H. Sun, J. Du and J. Li, J. Chem. Res., 2003, 157 CrossRef CAS.
- (a) V. McKee and S. S. Tandon, J. Chem. Soc., Dalton Trans., 1991, 221 RSC; (b) W.-X. Zhang, C.-Q. Ma, X.-N. Wang, Z.-G. Yu, Q.-J. Lin and D. H. Jiang, Chin. J. Chem., 1995, 13, 497 CrossRef CAS PubMed; (c) R. Shakya, P. H. Keyes, M. J. Heeg, A. Moussawel, P. A. Heiney and C. N. Verani, Inorg. Chem., 2006, 45, 7587 CrossRef CAS PubMed; (d) R. Shakya, S. S. Hindo, L. Wu, S. Ni, M. Allard, M. J. Heeg, S. R. P. da Rocha, G. T. Yee, H. P. Hratchian and C. N. Verani, Chem.–Eur. J., 2007, 13, 9948 CrossRef CAS PubMed; (e) S. S. Hindo, R. Shakya, R. Shanmugam, M. J. Heeg and C. N. Verani, Eur. J. Inorg. Chem., 2009, 4686 CrossRef CAS PubMed; (f) P. Roy, M. Nandi, M. Manassero, M. Ricco, M. Mazzani, A. Bhaumik and P. Banerjee, Dalton Trans., 2009, 9543 RSC; (g) M. Sarkar, R. Clerac, C. Mathoniere, N. G. R. Hearns, V. Bertolasi and D. Ray, Inorg. Chem., 2010, 49, 6575 CrossRef CAS PubMed; (h) M. Sarkar, R. Clerac, C. Mathoniere, N. G. R. Hearns, V. Bertolasi and D. Ray, Inorg. Chem., 2011, 50, 3922 CrossRef CAS PubMed; (i) D. Das, A. Guha, S. Das, P. Chakraborty, T. K. Mondal, S. Goswami and E. Zangrando, Inorg. Chem. Commun., 2012, 23, 113 CrossRef CAS PubMed; (j) M. S. Jana, S. Dey, J. L. Priego and R. Jimemez-Aparicio, Polyhedron, 2013, 59, 101 CrossRef CAS PubMed; (k) A. K. Ghosh and D. Ray, Polyhedron, 2013, 52, 370 CrossRef CAS PubMed; (l) S. Halder, S. Dey, C. Rizzoli and P. Roy, Polyhedron, 2014, 78, 85 CrossRef CAS PubMed; (m) S. Das, L. Sorace, A. Guha, R. Sanyal, H. Kara, A. Caneschi, E. Zangrando and D. Das, Eur. J. Inorg. Chem., 2014, 2753 CrossRef CAS PubMed; (n) M. Pait, E. Colacio and D. Ray, Polyhedron, 2015, 88, 90 CrossRef CAS PubMed.
- (a) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC; (b) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801 RSC; (c) X. Rong and C. Chunxia, Prog. Chem., 2012, 24, 1519 Search PubMed.
- N. Ikpo, C. Hoffmann, L. N. Dawe and F. M. Kerton, Dalton Trans., 2012, 6631 Search PubMed.
- (a) B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 15337 RSC; (b) X. You, Z. Wei, H. Wang, D. Li, J. Liu, B. Xu and X. Liu, RSC Adv., 2014, 4, 61790 RSC; (c) L. Wu, W. Zhong, B. Xu, Z. Wei and X. Liu, Dalton Trans., 2015, 8013 RSC.
- CrysAlisPro Softwarefor CCD diffractometers, Agilent Technologies, 2012 Search PubMed.
- SAINT and SMART software for CCD diffractometers, Bruker AXS Inc., Madison, USA, 2000 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, C34 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1040530–1040539. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra08696e |
| This journal is © The Royal Society of Chemistry 2015 |