
A green synthesis of highly substituted 1,5-diketones†
Abstract
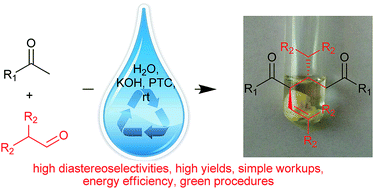
Highly substituted 1,5-diketones have been synthesized in water via the reactions between aryl methyl ketones and aldehydes and the subsequent dimerizations. The reaction was catalyzed by aqueous KOH. The advantages of these aqueous reactions over organic-solvent mediated reactions are better yields, better diastereoselectivities, faster reaction rates, simpler workups, and being more energy efficient. The reaction can be scaled up to 13.9 gram scale and the aqueous KOH can be reused for five cycles. A possible mechanism is proposed to explain the high diastereoselectivities.
 Please wait while we load your content...
Please wait while we load your content...

