
4-Nitro-2,1,3-benzoxadiazole derivatives as potential fluorescent sigma receptor probes†
 a
and
a
and
Abstract
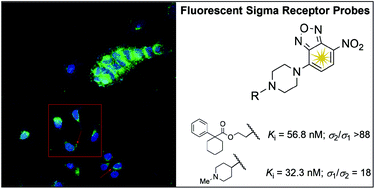
New fluorescent derivatives for σ receptors were designed and synthesized. To achieve this purpose, a 4-nitro-2,1,3-benzoxadiazole fluorescent tag was connected through a piperazine linker to a modified skeleton derived from selected σ receptor agonists or antagonists. Compounds 5g, 7b, 7e and 7g displayed high σ1 affinity and low σ1/σ2 selectivity (Kiσ1 ranging from 31.6 nM to 48.5 nM, Kiσ1/σ2 = 5–18), while compound 5d exhibited high σ2 affinity and selectivity (Kiσ2 = 56.8 nM, Kiσ1 > 5000 nM). Binding affinity studies revealed that compounds 5d, 5g, 7b, 7e and 7g showed no affinity towards several receptors including opioid, dopaminergic, serotonergic, adrenergic, muscarinic, histaminergic, N-methyl-D-aspartate (NMDA), NMDA receptor channel, or dopamine and serotonine transporters. The fluorescent properties, cellular uptake and confocal microscopy studies on 5d suggest a potential use of this probe to further clarify the molecular role of σ2 receptor subtypes in normal and cancer cells.
 Please wait while we load your content...
Please wait while we load your content...

