
Asymmetric reduction of imines with trichlorosilane catalyzed by valine-derived formamide immobilized onto magnetic nano-Fe3O4†
Xin Geab,
Chao Qiana,
Xiaoming Yea and
Xinzhi Chen*a
aKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, P.R China. E-mail: xzchen@zju.edu.cn; Tel: +86-571-87951615
bSchool of Chemical and Material Engineering, Jiangnan University, P.R China
First published on 27th July 2015
Abstract
Magnetic nano-Fe3O4-supported organocatalysts were synthesized by anchoring valine-derived formamide onto the surface of Fe3O4 magnetic nanoparticles, which were applied in the asymmetric reduction of imines with trichlorosilane at room temperature in toluene. The high level of yield and enantioselectivity catalyzed by magnetic nano-Fe3O4-supported organocatalysts was obtained. In the immobilization process, CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) “click chemistry” was used as the anchored bridge. The magnetic nanoparticles can simplify the recovery of the organocatalyst and its separation from the reaction system. By an external magnet, the catalyst can be recycled and reused five times without a remarkable activity decline.
Introduction
In addition to organometallic and bio-catalytic catalysts, as small organic molecules, organocatalysts are capable of promoting asymmetric synthesis and have rapidly gained much attention due to their relatively easy operation and environmentally friendly nature. Thus, for the asymmetric reduction of imine, an organocatalytic approach is an attractive alternative method to obtain chiral amine. Moreover, the Lewis-basic organocatalytic reduction with trichlorosilane is an emerging organocatalytic methodology. After Matsumura et al. firstly reported the N-formyl-L-proline anilide as a Lewis-basic organocatalyst to catalyze the reduction of imines with trichlorosilane in moderate enantioselectivity,1 some highly effective Lewis-basic organocatalysts were developed to catalyze the asymmetric reduction of imines. Besides N-formyl-L-proline anilide, some other L-proline-derived organocatalysts, such as N-picolinoyl-L-pyrrolidine,2 L-proline derived C2-symmetric chiral tetraamide,3 N-formyl proline fatty amide4 and N-substituted prolines,5 were also developed and could successfully catalyze the asymmetric reduction of imines. Then Malkov and Kocovsky6–8 discovered that N-formyl-L-valine derived Lewis-basic organocatalysts could significantly improve enantioselectivity. L-pyrrolidine and L-valine have become important components of Lewis-basic organocatalysts for the asymmetric reduction of imines. Sun et al. exploited pipecolinic acid derived,9–11 piperazine-2-carboxylic acid derived12 and S-chiral13–15 organocatalysts to reduce imines with trichlorosilane in high enantioselectivity. Recently, we described carbohydrate-derived pyridinecarboxylic organocatalysts for the enantioselective reduction of imines with trichlorosilane in high yield (up to 93%) and with moderate enantioselectivity (up to 75%).16 In sum, considering the extensive source and catalytic effective, N-formyl-L-valine derived Lewis-basic organocatalysts are more successful organocatalysts for the asymmetric reduction of imine.However, for typical organocatalytic procedures, the problems about separation and recovery of catalyst from the reaction medium arose. To overcome the separation and recovery difficulties, the common solution is the immobilization of organocatalyst. In 2009, Malkov and Kocovsky first developed N-formyl-L-valine derived organocatalysts immobilised onto gold nanoparticles.17 Then, they prepared soluble polymer-supported valine derived organocatalyst, which could be recovered through pouring into the methanol and reused at least five times without loss of activity.18 In 2010, they synthesized dendron-anchored N-formyl-L-valine derived organocatalysts to catalyze the symmetric reduction of imines with trichlorosilane.19 The dendron-anchored organocatalysts can be removed by precipitation and centrifugation. As supported carriers, magnetic nanoparticles (MNPs) have been attracted much attention for their simple and easy recover from the liquid-phase reaction by magnetically derived separation.20–27 Moreover, MNPs supported catalyst can rapidly disperse in reaction solvent. Thus, MNPs have been applied in traditional metal catalysts, organocatalysts, and even enzyme. Recently, MNPs supported chiral organocatalysts and ionic liquid have been successfully prepared to catalyze Michael addition,28,29 ammoximation,30 Suzuki–Miyaura coupling reaction31 and so on. To the best of our knowledge, N-formyl-L-valine derived organocatalyst immobilized onto magnetic nano-Fe3O4 was not reported. Herein, we present magnetic nano-Fe3O4-supported N-formyl-L-valine derived organocatalyst by click reaction and its application in asymmetric reduction of imines with trichlorosilane.
Results and discussion
Preparation and characterization of the catalyst
The synthesis of the precursor N-formyl-L-valine derived organocatalyst 4 was commenced with the methylation of N-BOC-L-valine with iodomethane in presence of neat sodium hydride,32,33 which afforded the corresponding N-methylated product 1. Then the precursor N-formyl-L-valine derived organocatalyst 4 was prepared by the condensation of the BOC-protected N-methyl valine 1 with respective aniline derivative 2, followed by BOC-deprotection with CF3COOH and N-formylation.34Magnetic nano-Fe3O4 particles was prepared via the coprecipitation of ferric chloride hexahydrate and ferrous chloride tetrahydrate in an alkaline medium.35 Considering the aggregation of magnetic nano-Fe3O4 particles (MNPs) and further modification on the surface, it is a good solution that the magnetic nanoparticles was coated with silica and the silica-coated Fe3O4 (SiO2@Fe3O4) was prepared according to the literature procedures.26,36 The abundant surface hydroxyl groups of MNPs (SiO2@Fe3O4) were used as carriers to graft 3-azidopropyl moiety onto surface with 3-azidopropyltrimethoxysilane.21 Finally, the catalytically active species 4 was immobilized onto azide@MNPs using CuI-catalyzed azide-alkyne 1,3-dipolar cycloaddition (CuAAC) “click chemistry”33,37–40 and the desired MNPs-supported organocatalyst 5 was obtained. CuAAC reaction was used as an anchored bridge (Scheme 1).
 | ||
| Scheme 1 Synthese of N-formyl-L-valine derived organocatalyst supported onto MNPs. | ||
The XRD patterns of SiO2@Fe3O4 and MNPs-supported organocatalyst 5 were shown in Fig. 1 and matched well with the date of the standard sample.30 It exhibits diffraction peaks of crystal planes (220), (311), (400), (422), (511), (440) and relative intensity. Comparing with the XRD patterns of SiO2@Fe3O4 and MNPs-supported organocatalyst 5, there is no significant influence on the structure of Fe3O4 nanoparticles in the immobilization of valine-derived formamide catalyzed by CuAAC click chemistry. However, the broad peak of amorphous silica phase in the shell of the silica-coated Fe3O4 from 2θ = 20° to 30° is not obvious.22 Moreover, the transmission electron microscopy (TEM) images of SiO2@Fe3O4, azide@MNPs and MNPs-supported organocatalyst 5 are shown in Fig. 2. The thickness of the silica shell surrounding the bare nano-Fe3O4 particles is about 3–5 nm and the average particle diameter is 20–30 nm (in Fig. 2a). The TEM morphologies of azide@MNPs and MNPs-supported organocatalyst 5 were similar to that of the SiO2@Fe3O4 and the core–shell structures were obvious (in Fig. 2b–c).
 | ||
| Fig. 1 XRD patterns of SiO2@Fe3O4 (a) and MNPs-supported organocatalyst 5 (b). | ||
 | ||
| Fig. 2 TEM images of SiO2@Fe3O4 (a), azide@MNPs (b), MNPs-supported organocatalyst 5 (c) and MNPs-supported organocatalyst 5 after being reused five times (d). | ||
In order to make sure whether the functionalization of azide@MNPs with valine-derived formamide catalyzed by CuAAC click chemistry is successful, the Fourier transform infrared spectroscopy (FT-IR) analysis was employed to further investigate. As shown in Fig. 3, the spectrum of all the samples exhibited broad peaks at around 3400 and 1640 cm−1, which were attributed to the OH group on the silica surface and adsorbed water, respectively. Meanwhile, the peaks at 1080 and 575 cm−1 should be due to Si–O–Si stretch vibration and Fe–O vibration modes, respectively. For azide@MNPs, azido group was introduced into SiO2@Fe3O4 and led to a characteristic peak at 2100 cm−1, following the appearance of typical bands at 2940, 2850 and 1390 cm−1 (alkyl chain stretching and deformation vibrations). The loading amount of azido was determined to be 0.28 mmol g−1 by elemental analysis. After the successful functionalization of azide@MNPs with valine-derived formamide catalyzed by CuAAC click chemistry, the strong adsorption peak of azido group was weakened. Moreover, new band at 1560 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC vibrations) emerged in the formation of the 1,2,3-triazole linker. The loading of the N-formyl-L-valine derived organocatalyst was determined to be 0.24 mmol g−1 by elemental analysis.
N and CC vibrations) emerged in the formation of the 1,2,3-triazole linker. The loading of the N-formyl-L-valine derived organocatalyst was determined to be 0.24 mmol g−1 by elemental analysis.
 | ||
| Fig. 3 FI-IR spectrum of SiO2@Fe3O4 (a), azide@MNPs (b) and MNPs-supported organocatalyst 5 (c). | ||
The magnetism of SiO2@Fe3O4 and MNPs-supported organocatalyst 5 was investigated at room temperature. As shown in Fig. 4, the two samples can be completely saturated at high fields of up to 2.0 T. When active species 4 was grafted on the surface of SiO2@Fe3O4, the saturated magnetism was decreased from the 59.7 to 24.5 emu g−1. Thus, the efficient magnetization of MNPs-supported organocatalyst 5 could guarantee simple separation with an external magnet.
 | ||
| Fig. 4 Magnetic curves of SiO2@Fe3O4 (a) and MNPs-supported organocatalyst 5 (b). | ||
The stability of the MNPs-supported organocatalyst 5 was investigated by the thermogravimetric (TG) analysis. As the Fig. 5 shown, an initial weight loss up to 160 °C was detected owing to the remove of adsorbed water on the support. Thermal degradation of MNPs-supported organocatalyst 5 occurred from 160 °C to 575 °C, which resulted into the weight loss of 13%. It revealed that the catalyst exhibited the excellent stability. Moreover, the decomposition of the organocatalyst was mainly occurred from 500 °C to 575 °C. Thus, the MNPs-supported organocatalyst 5 is stable around or below 160 °C.
 | ||
| Fig. 5 TG-DTG analysis for azide@MNPs. | ||
Catalytic activity of MNPs-supported organocatalyst in asymmetric reduction of imines with trichlorosilane
With the MNPs-supported organocatalyst 5 in hand, we evaluated its catalytic activity in enantioselective reduction of imines with trichlorosilane. The asymmetric reduction of imine 6a with trichlorosilane was selected as model reaction. Initially, the reaction conditions were optimized and the results were shown in Table 1. We first attempted the active species 4 to catalyze asymmetric reduction of imine 6a, which affording 95% yield and 89% ee separately with the (S)-enantiomer (Table 1, entry 1). When the MNPs-supported organocatalyst 5 was used for this reduction, 94% yield and 85% ee could be obtained (Table 1, entry 2). By contrast, it was obvious that the catalytic activity did not decline. The reactivity of the immobilized catalyst in different solvent was studied (Table 1, entries 2–5). In the asymmetric reduction of imines catalyzed by N-formyl-L-valine derived organocatalyst, the hydrogen bonding and the π–π interactions influenced the enantioselectivity.33 Thus, toluene is the best solvent, which takes both in consideration. When the reaction was carried out in chloroform and dichloromethane, the catalytic activity declined slightly. Moreover, in acetonitrile, the ee value is only 24%.
|
||||
|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Yieldb (%) | eec,d (%) (config) |
| a The reactions were carried out with catalyst and 1.5 equiv. of SiHCl3 on a 0.5 mmol scale in 2.0 mL of solvent for 24 h.b Isolated yield based on the imine.c Determined by chiral HPLC.d The absolute configuration was determined by HPLC via comparison with authentic samples.6,12 | ||||
| 1 | 4 (10) | Toluene | 95 | 89 (S) |
| 2 | 5 (10) | Toluene | 94 | 85 (S) |
| 3 | 5 (10) | CH2Cl2 | 93 | 74 (S) |
| 4 | 5 (10) | CHCl3 | 91 | 81 (S) |
| 5 | 5 (10) | CH3CN | 89 | 24 (S) |

Having testing the activity of the MNPs-supported organocatalyst 5 for the reduction of imine 6a, the substrate scope of MNPs-supported organocatalyst 5 was explored further. The results were summarized in Table 2. As the substrate 6a of the model reaction, the product was obtained in 94% yield and 85% ee (Table 2, entry 1). For aromatic N-Ph imines 6b–6e, satisfactory yields and enantioselectivities were affording (Table 2, entry 2–5). The imines 6e with electron-donating group showed lower catalytic activity than that of 6a, affording 72% yield and 67% ee. The aromatic N-Ph imines 6b–6d with electron-withdrawing groups exhibited more excellent asymmetric reduction.
|
|||||
|---|---|---|---|---|---|
| Entry | Imine | R1 | R2 | Yield (%)b | ee (%)c |
| a The reactions were carried out with 10 mol% catalyst 5 and 1.5 equiv. of SiHCl3 on a 0.5 mmol scale in 2.0 mL of toluene for 24 h.b Isolated yield based on the imine.c Determined by chiral HPLC. | |||||
| 1 | 6a | C6H5 | C6H5 | 94 | 85 |
| 2 | 6b | 4-FC6H4 | C6H5 | 95 | 88 |
| 3 | 6c | 4-ClC6H4 | C6H5 | 97 | 91 |
| 4 | 6d | 4-NO2C6H4 | C6H5 | 94 | 92 |
| 5 | 6e | 4-CH3C6H4 | C6H5 | 72 | 67 |
| 6 | 6f | C6H5 | 4-FC6H4 | 94 | 81 |
| 7 | 6g | C6H5 | 4-ClC6H4 | 93 | 83 |
| 8 | 6h | C6H5 | 3-ClC6H4 | 84 | 74 |
| 9 | 6i | C6H5 | 3-BrC6H4 | 92 | 86 |
| 10 | 6j | C6H5 | 4-CH3C6H4 | 81 | 52 |
| 11 | 6k | C6H5 | 2-CH3C6H4 | 67 | 25 |

The effect of the imine N-substituent (R2) is similar to that of R1, affording the 25–86% ee value (Table 2, entries 6–12). The electron-poor N-substituent imines exhibited higher catalytic activity than electron-rich imines. The electron-poor N-substituent imines 6f–6i could be reduced in 84–94% yield and 74–86% ee (Table 2, entries 6–9). For electron-rich N-substituent imines 6j–6k, the yields and enantioselectivities were 67–81% and 25–52% separately (Table 2, entries 10 and 11).
Reuse of MNPs-supported organocatalyst
The main advantage of MNPs is easy recover from the liquid-phase reaction by magnetically derived separation. Thus, we attempted to study the possibility of reuse of the MNPs-supported organocatalyst 5 in the model reaction of imine 6a asymmetric reduction. The catalyst 5 could be readily dispersed in the reaction media. Once the reaction completed, the catalyst was easily recovered from the reaction system by external magnet, then washed by ethanol and dried under vacuum. The recovered catalyst could be reused and easily redispersed in reaction medium. Generally, the recycling catalyst was less than the previous (see in the ESI†). The slight loss of catalyst led to slight decrease of the yield and enantioselectivity. After five cycles, the catalyst had not an obvious loss of catalytic activity (Fig. 6). As shown in Fig. 2d, a TEM of the recovered catalyst was observed. The morphology and size have no obvious changes. It indicated that the MNPs-supported organocatalyst 5 could be reused and stable.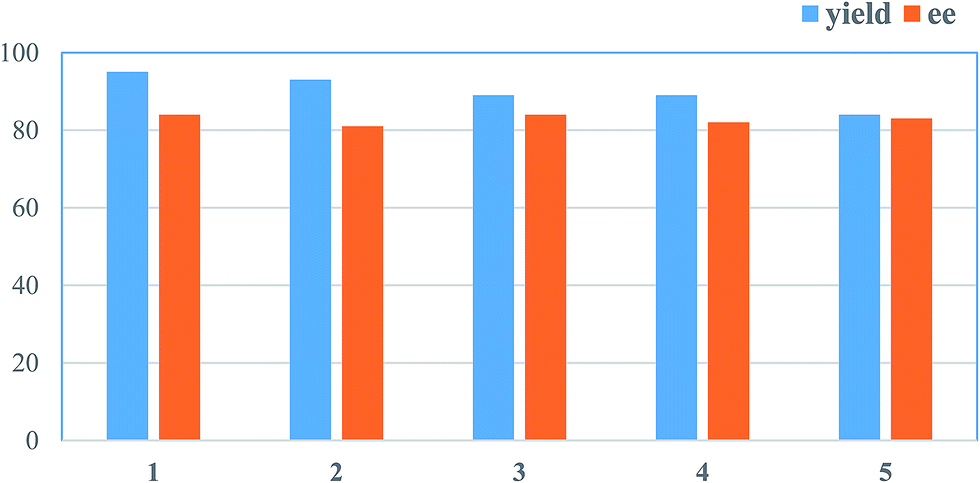 | ||
| Fig. 6 Recycling and reuse of catalyst 5. a The reactions were carried out with 10 mol% catalyst 5 and 1.5 equiv. of SiHCl3 on a 0.5 mmol scale in 2.0 mL of toluene for 24 h. b Isolated yield based on the imine. c ee value was determined by chiral HPLC. | ||
Conclusion
In sum, we achieved successfully the immobilization of valine-derived formamide organocatalyst onto magnetic nano-Fe3O4 by click reaction. The obtained catalyst exhibited high catalytic activity in the asymmetric reduction of imines with trichlorosilane. Furthermore, the catalyst could be readily recovered by an external magnet and reused five times without an obvious activity decline.Acknowledgements
The authors are grateful for the financial support from the Natural Science Foundation of China (21376213, 21476194), the Research Fund for the Doctoral Program of Higher Education of China (20120101110062) and the Zhejiang Provincial Public Technology Research of China (2014C31123).References
- F. Iwasaki, O. Onomura, K. Mishima, T. Kanematsu, T. Maki and Y. Matsumura, Tetrahedron Lett., 2001, 42, 2525–2527 CrossRef CAS.
- O. Onomura, Y. Kouchi, F. Iwasaki and Y. Matsumura, Tetrahedron Lett., 2006, 47, 3751–3754 CrossRef CAS PubMed.
- Z. Y. Wang, S. Y. Wei, C. Wang and J. Sun, Tetrahedron: Asymmetry, 2007, 18, 705–709 CrossRef CAS PubMed.
- Z. G. Zhang, P. Rooshenas, H. Hausmann and P. R. Schreiner, Synthesis, 2009, 1531–1544 CAS.
- T. Kanemitsu, A. Umehara, R. Haneji, K. Nagata and T. Itoh, Tetrahedron, 2012, 68, 3893–3898 CrossRef CAS PubMed.
- A. V. Malkov, A. Mariani, K. N. MacDougall and P. Kocovsky, Org. Lett., 2004, 6, 2253–2256 CrossRef CAS PubMed.
- A. V. Malkov, S. Stoncius, K. N. MacDougall, A. Mariani, G. D. McGeoch and P. Kocovsky, Tetrahedron, 2006, 62, 264–284 CrossRef CAS PubMed.
- A. V. Malkov, K. Vrankova, R. C. Sigerson, S. Stoncius and P. Kocovsky, Tetrahedron, 2009, 65, 9481–9486 CrossRef CAS PubMed.
- Z. Y. Wang, X. X. Ye, S. Y. Wei, P. C. Wu, A. J. Zhang and J. Sun, Org. Lett., 2006, 8, 999–1001 CrossRef CAS PubMed.
- L. Zhou, Z. Y. Wang, S. Y. Wei and J. Sun, Chem. Commun., 2007, 2977–2979 RSC.
- Z. Y. Wang, C. Wang, L. Zhou and J. Sun, Org. Biomol. Chem., 2013, 11, 787–797 CAS.
- Z. Y. Wang, M. Cheng, P. C. Wu, S. Y. Wei and J. Sun, Org. Lett., 2006, 8, 3045–3048 CrossRef CAS PubMed.
- D. Pei, Z. Wang, S. Wei, Y. Zhang and J. Sun, Org. Lett., 2006, 8, 5913–5915 CrossRef CAS PubMed.
- D. Pei, Y. Zhang, S. Y. Wei, M. Wang and J. Sun, Adv. Synth. Catal., 2008, 350, 619–623 CrossRef CAS PubMed.
- C. Wang, X. Wu, L. Zhou and J. Sun, Chem.–Eur. J., 2008, 14, 8789–8792 CrossRef CAS PubMed.
- X. Ge, C. Qian, Y. B. Chen and X. Z. Chen, Tetrahedron: Asymmetry, 2014, 25, 596–601 CrossRef CAS PubMed.
- A. V. Malkov, M. Figlus, G. Cooke, S. T. Caldwell, G. Rabani, M. R. Prestly and P. Kocovsky, Org. Biomol. Chem., 2009, 7, 1878–1883 CAS.
- A. V. Malkov, M. Figlus, M. R. Prestly, G. Rabani, G. Cooke and P. Kocovsky, Chem.–Eur. J., 2009, 15, 9651–9654 CrossRef CAS PubMed.
- M. Figlus, S. T. Caldwell, D. Walas, G. Yesilbag, G. Cooke, P. Kocovsky, A. V. Malkov and A. Sanyal, Org. Biomol. Chem., 2010, 8, 137–141 CAS.
- T. Y. Cheng, D. C. Zhang, H. X. Li and G. H. Liu, Green Chem., 2014, 16, 3401–3427 RSC.
- P. Riente, C. Mendoza and M. A. Pericas, J. Mater. Chem., 2011, 21, 7350–7355 RSC.
- Q. W. Du, W. Zhang, H. Ma, J. Zheng, B. Zhou and Y. Q. Li, Tetrahedron, 2012, 68, 3577–3584 CrossRef CAS PubMed.
- A. G. Hu, S. Liu and W. B. Lin, RSC Adv., 2012, 2, 2576–2580 RSC.
- H. J. Xu, X. Wan, Y. Y. Shen, S. Xu and Y. S. Feng, Org. Lett., 2012, 14, 1210–1213 CrossRef CAS PubMed.
- L. Yu, M. Wang, P. H. Li and L. Wang, Appl. Organomet. Chem., 2012, 26, 576–582 CrossRef CAS PubMed.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Green Chem., 2012, 14, 201–208 RSC.
- D. Singh, H. Chandra and V. Krishna, Separ. Sci. Tech., 2015, 50, 437–445 CrossRef CAS PubMed.
- A. G. Ying, S. Liu, Y. X. Ni, F. L. Qiu, S. L. Xu and W. Y. Tang, Catal. Sci. Technol., 2014, 4, 2115–2125 CAS.
- A. G. Ying, F. L. Qiu, C. L. Wu, H. A. Hu and J. G. Yang, RSC Adv., 2014, 4, 33175–33183 RSC.
- L. W. Tong Liu, H. Wan and G. Guan, Catal. Commun., 2014, 49, 20–24 CrossRef PubMed.
- Q. Zhang, H. Su, J. Luo and Y. Y. Wei, Catal. Sci. Technol., 2013, 3, 235–243 CAS.
- A. V. Malkov, K. Vrankova, M. Cerny and P. Kocovsky, J. Org. Chem., 2009, 74, 8425–8427 CrossRef CAS PubMed.
- X. Ge, C. Qian and X. Z. Chen, Tetrahedron: Asymmetry, 2014, 25, 1450–1455 CrossRef CAS PubMed.
- A. V. Malkov, M. Figlus, S. Stoncius and P. Kocovsky, J. Org. Chem., 2007, 72, 1315–1325 CrossRef CAS PubMed.
- A. Bee, R. Massart and S. Neveu, J. Magn. Magn. Mater., 1995, 149, 6–9 CrossRef CAS.
- X. L. Zhang, P. H. Li, Y. Ji, L. Zhang and L. Wang, Synthesis, 2011, 2975–2983 CAS.
- L. Ji, G. Q. Zhou, C. Qian and X. Z. Chen, Eur. J. Org. Chem., 2014, 2014, 3622–3636 CrossRef CAS PubMed.
- K. D. Hanni and D. A. Leigh, Chem. Soc. Rev., 2010, 39, 1240–1251 RSC.
- J. M. Holub and K. Kirshenbaum, Chem. Soc. Rev., 2010, 39, 1325–1337 RSC.
- C. le Droumaguet, C. Wang and Q. Wang, Chem. Soc. Rev., 2010, 39, 1233–1239 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08516k |
| This journal is © The Royal Society of Chemistry 2015 |