DOI:
10.1039/C5RA08496B
(Paper)
RSC Adv., 2015,
5, 57917-57929
Benzo[α]phenoxazines and benzo[α]phenothiazine from vitamin K3: synthesis, molecular structures, DFT studies and cytotoxic activity†
Received
8th May 2015
, Accepted 26th June 2015
First published on 26th June 2015
Abstract
Synthesis and characterization of fluorescent benzo[α]phenoxazines viz., M-1B (10-chloro-6-methyl-7a,11a-dihydro-5H-benzo[α]phenoxazin-5-one), M-2B 6,10-dimethyl-7a,11a-dihydro-5H-benzo[α]phenoxazin-5-one), M-3B (6-methyl-7a,11a-dihydro-5H-benzo[α]phenoxazin-5-one) and benzo[α]phenthiazine, M-4B (6-methyl-5H-benzo[α]phenothiazin-5-one) were carried out. 1H and 13C chemical shifts were assigned from the 2DgHSQCAD NMR experiments. Compound M-1B crystallizes in the orthorhombic space group P212121, while M-2B and M-4B crystallize in the monoclinic space group P21/c. The crystal network of M-1B showed slipped π–π stacking and Cl⋯Cl interactions, while M-2B facilitated ladder like π–π stacked polymeric chains. The C⋯S contacts were observed in the crystal environment of M-4B. All these structures possess C–H⋯O interactions. Electronic structure and charge distribution in terms of molecular electrostatic potential and frontier orbital analyses based on the MO6-2X based density functional theory further showed that monomer and dimer structures are in keeping with the single crystal X-ray data and provide insights for the growth of the crystal network. Antiproliferative activity of M-1B–M-4B was determined from the MTT assay against a human breast adenocarcinoma cell line (MCF-7), human carcinoma cell line (HeLa) and normal skin cell line. All these compounds showed significant cytotoxic activity against MCF-7 and HeLa by inducing apoptosis and thus can be viewed as potential candidates for antitumor therapy. Compounds M-2B and M-4B were further found to be topoisomerase II inhibitors.
Introduction
Benzo[α]phenoxazines are fluorescent molecules with a wide range of pharmacological applications.1 Phenoxazine derivatives viz., benzo[α]phenoxzinium salts, show absorption maxima higher than 600 nm and the strong red fluorescence thereof facilitates their use as long wavelength fluorophores.2,3 Preferred for biological applications4–9 these encompass labelling of small synthetic molecules as well as large biomolecules including proteins, antibodies or nucleotides in DNA studies. Phenoxazines possessing antiproliferative,10 antitumor,11 anti-inflammatory,12 anti-tuberculosis13 and multidrug resistance activities14 prevent human amyloid disorders15 and protect neuronal cells from death by oxidative stress.16,17 Furthermore, it was shown that the use of phenothiazine derivatives belonging to neuroleptic drugs,18 reduce tumor-antigen expression19 and cytotoxicity against myelogeneous leukemia cell lines.20 The underlying mechanism for antiproliferative activity of phenoxazine derivatives stem from DNA intercalative binding facilitated via noncovalent interactions or π–π stacking21,22 which render stability to these planar polycycles. Alternatively phenoxazine systems engender free radical intermediates leading to oxidative stress.23 Some of these phenoxazine derivatives displayed apoptotic activity against different cell lines, both in caspase dependent and independent manner.24,25 Fluorescent heterocycle of phenoxazines are valuable for staining nucleic acids in solutions, electrophoretic gels, matrices, blotting experiments and assays employing intact, live cells.26 Furthermore, modifications of phenoxazine moiety yield compounds those directly bind to proteins through hydrophobic interactions which subsequently are utilized in monitoring protein conformation alterations or for therapeutic purposes.
Topoisomerase are the enzymes those control the DNA topology, a requisite for division or proliferation of cells. These enzymes are important target for cancer chemotherapy and assist numerous DNA transactions through a complex series of DNA strand breakage and rejoining reactions. Topoisomerases resolves the entanglement problems generated during DNA replication, repair and transcription processes. Depending on the DNA breaks there are two types of topoisomerases; Topo I, which break single strand DNA, while Topo II generate double strand DNA breaks.27,28 Topo II enzymes plays key role in DNA metabolism and chromosome structure29 and it is the primary cytotoxic target for the potent anticancer drugs, which include anthracyclines, acridines and epipodo phyllotoxins.30 Depending on the mode of action the topoisomerase II inhibitors are classified as (i) topoisomerase II poisons, which involves in breaking–rejoining reaction of the enzyme that involves trapping of covalent reaction intermediates, usually referred as the cleaved complexes, or (ii) catalytic inhibitors inhibiting the activity of topoisomerase II prior to the formation of the cleaved complex.31 Heteroatom planar polycyclic compounds are used in cancer chemotherapy as topoisomerase inhibitor/poison.32–39 DNA intercalation has been believed to be a prefer mode of action for such compounds.
In present endeavour, benzo[α]phenoxazines were synthesized by reacting vitamin K3 (menadione, 2-methyl-1,4-naphthoquinone) with 2-aminophenols (M-1B to M-3B) and 2-aminothiophenol (M-4B) derivatives. Molecular structures of M-1B, M-3B and M-4B have been studied by single crystal X-ray diffraction studies, which revealed their π–π stacking abilities. The structural investigations were combined with those from the density functional based calculations. The antiproliferative activity against human carcinoma HeLa cell line, drug uptake and topoisomerase II inhibitor activity of these compounds have further been investigated.
Results and discussion
Synthesis
Benzo[α]phenoxazine derivatives were synthesized by condensation of vitamin K3 (1) with 4-R-2-aminophenols (R = Cl = M-1B, CH3 = M-2B, H = M-3B) refluxed in methanol, while M-4B was obtained at room temperature (26 °C) by reacting vitamin K3 and 2-aminothiophenol (Scheme 1). 6-Methyl-5H-benzo[α]phenoxazine-5-one and 6-methyl-5H-benzo[α]phenothiazin-5-one (M-3B and M-4B respectively in present investigation) were previously synthesized by reactions of alkyl radicals on corresponding 5H-benzo[α]phenoxazine-5-one40 and 5H-benzo[α]phenothiazin-5-one41 respectively.
 |
| | Scheme 1 General reaction scheme for synthesis of M-1B to M-4B. | |
FT-IR, UV-visible, fluorescence, 1H and 13C NMR studies
The νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O frequency in all compounds was assigned at 1629 ± 1 cm−1, while νCN stretching frequency42–45 was assigned at 1585 ± 10 cm−1 in the FT-IR spectra except in M-1B it was observed at 1574 cm−1 (Fig. S22 and Table S1 in ESI†). The para-naphthoquinone (p-NQ) vibration10 of quinonoid ring (∼1260 cm−1) was absent in all compounds however a peak due to νC–O was observed ∼1240 cm−1 in M-1B, M-3B and M-4B and ∼1226 cm−1 in M-2B. vC–Cl stretching frequency was assigned to 736 cm−1 in M-1B.
O frequency in all compounds was assigned at 1629 ± 1 cm−1, while νCN stretching frequency42–45 was assigned at 1585 ± 10 cm−1 in the FT-IR spectra except in M-1B it was observed at 1574 cm−1 (Fig. S22 and Table S1 in ESI†). The para-naphthoquinone (p-NQ) vibration10 of quinonoid ring (∼1260 cm−1) was absent in all compounds however a peak due to νC–O was observed ∼1240 cm−1 in M-1B, M-3B and M-4B and ∼1226 cm−1 in M-2B. vC–Cl stretching frequency was assigned to 736 cm−1 in M-1B.
There were mainly two bands observed in UV-Visible spectra of parent vitamin K3 in DMSO at 266 nm and 330 nm and four bands to all compounds (Fig. 1). In M-1B to M-3B the band ∼330 nm (of parent MQ) showed shift ∼25 to 30 nm (∼355 nm). Two another bands observed in UV region ∼260 nm to 290 nm were assigned to π–π* transitions. The n–π* transition was observed ∼450 nm (M-1B to M-3B) which shifts to ∼480 nm in M-4B. A fluorescence emission band was observed between 450–700 nm. M-2B showed maximum fluorescent intensity than other compounds despite using the same concentrations in recording of the spectra.
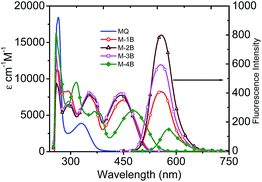 |
| | Fig. 1 UV-visible and fluorescence spectra of M-1B to M-4B in DMSO. | |
1H and 13C chemical shift were assigned to all compounds and are presented in Fig. 2.
 |
| | Fig. 2 1H and 13C NMR chemical shifts of M-1B to M-4B. | |
Single crystal X-ray diffraction studies of M-1B, M-2B and M-4B
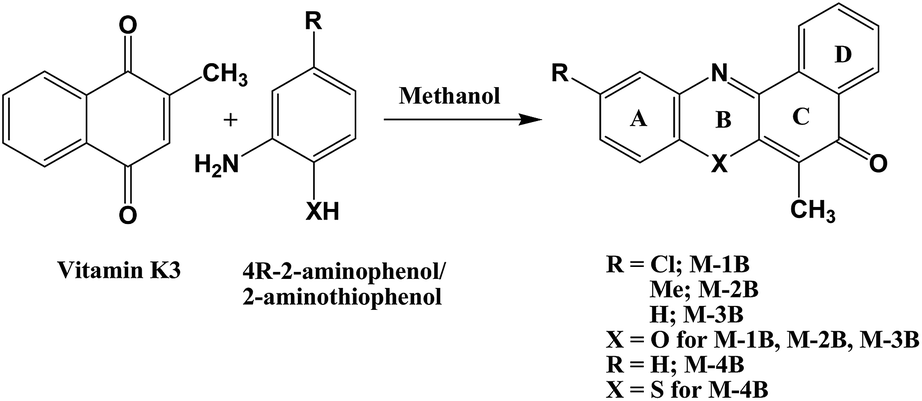
M-1B crystallizes in orthorhombic space group P212121, while M-2B and M-4B crystallize in monoclinic space group P21/c. ORTEP plots are presented in Fig. 3, and the crystallographic data is summarized in Table 1. All the crystallized compounds showed planar polycycles. The carbonyl distance C(2)–O(1) was observed as ∼1.23 Å in M-1B and M-2B,10,42–45 while it was longer in M-4B (1.246 Å). The bond distance of C(9)–N(10) was found to be ∼1.30 Å similar to the imino functional moiety (CN) of the naphthoquinoneoximes.46 The bond distances in all these crystalline systems usually by and large, match with those for quinonoid bond distances in ring B (Scheme 1).
 |
| | Fig. 3 ORTEP plots of M-1B, M-2B and M-4B. | |
Table 1 Crystallographic data of M-1B, M-2B and M-4B
| Identification code |
M-1B |
M-2B |
M-4B |
| Empirical formula |
C17H10ClNO2 |
C18H13NO2 |
C17H11NOS |
| Formula weight |
295.71 |
275.29 |
277.33 |
| Temperature |
100(2) K |
100(2) K |
100(2) K |
| Wavelength |
0.71073 Å |
1.54178 Å |
0.71073 Å |
| Crystal system |
Orthorhombic |
Monoclinic |
Monoclinic |
| Space group |
P212121 |
P21/c |
P21/c |
| Unit cell dimensions |
a = 4.7569(9) Å, b = 9.660(2) Å, c = 28.117(5) Å |
a = 7.1715(6) Å, b = 21.816(2) Å, β = 112.283(4) Å, c = 8.9610(9) Å |
a = 15.172(3) Å, b = 3.8385(8) Å, β = 107.902(9) Å, c = 22.105(4) Å |
| Volume |
1292.0(4) Å3 |
1297.3(2) Å3 |
1225.0(4) Å3 |
| Z |
4 |
4 |
4 |
| Density (calculated) |
1.520 mg m−3 |
1.410 mg m−3 |
1.504 mg m−3 |
| Absorption coefficient |
0.299 mm−1 |
0.742 mm−1 |
0.257 mm−1 |
| F(000) |
608 |
576 |
576 |
| Crystal size |
0.264 × 0.037 × 0.012 mm3 |
0.32 × 0.12 × 0.08 mm3 |
0.40 × 0.02 × 0.02 mm3 |
| Theta range for data collection |
3.028 to 30.210° |
4.053 to 67.383° |
2.724 to 28.999° |
| Index ranges |
−6 ≤ h ≤ 6, −13 ≤ k ≤ 13, −39 ≤ l ≤ 39 |
−7 ≤ h ≤ 8, −26 ≤ k ≤ 25, −10 ≤ l ≤ 10 |
−20 ≤ h ≤ 20, −5 ≤ k ≤ 5, −30 ≤ l ≤ 30 |
| Reflections collected |
35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 695 695 |
30049 |
15404 |
| Independent reflections |
3840 [R(int) = 0.0956] |
2298 [R(int) = 0.0581] |
3243 [R(int) = 0.0734] |
| Completeness to theta = 25.242° |
99.6% |
98.0% |
99.6% |
| Absorption correction |
Gaussian |
Gaussian |
Gaussian |
| Max. and min. transmission |
0.99706 and 0.95811 |
0.94601 and 0.84701 |
0.99571 and 0.94895 |
| Refinement method |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
Full-matrix least-squares on F2 |
| Data/restraints/parameters |
3840/0/191 |
2298/0/193 |
3243/0/182 |
| Goodness-of-fit on F2 |
1.040 |
1.059 |
1.116 |
| Final R indices [I > 2sigma(I)] |
R1 = 0.0397, wR2 = 0.0813 |
R1 = 0.0543, wR2 = 0.1482 |
R1 = 0.0657, wR2 = 0.1519 |
| R indices (all data) |
R1 = 0.0545, wR2 = 0.0870 |
R1 = 0.0660, wR2 = 0.1611 |
R1 = 0.0898, wR2 = 0.1659 |
| Extinction coefficient |
n/a |
0.0032(8) |
n/a |
| Largest diff. peak and hole |
0.314 and −0.378 e Å−3 |
0.295 and −0.297 e Å−3 |
0.720 and −0.611 e Å−3 |
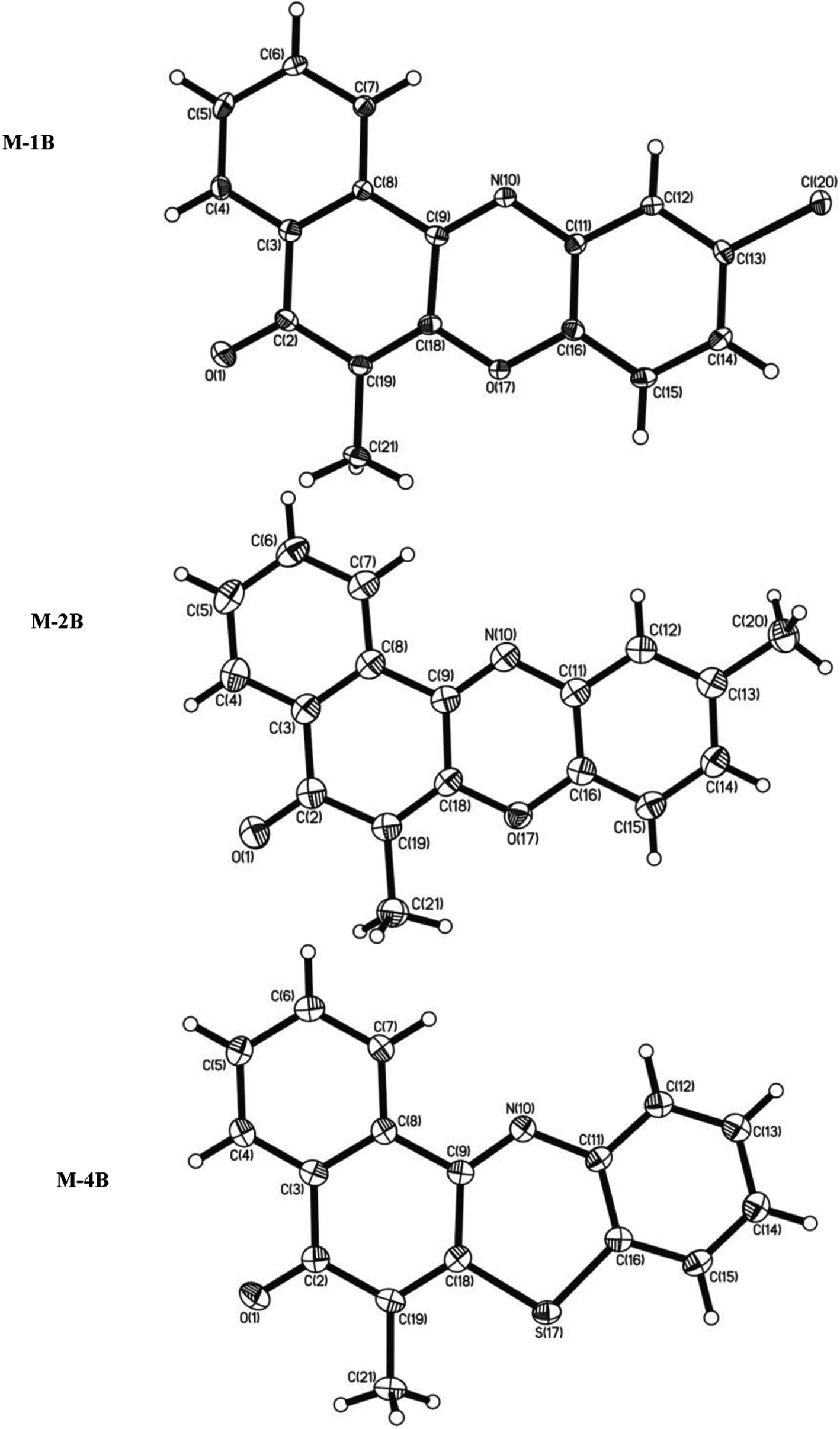
Each M-1B molecule was in vicinity to eight neighbouring molecules (Fig. S23†) via C–H⋯O, C–H⋯Cl, Cl⋯Cl (Table 2) and π–π stacking interactions. Slipped π–π stacking were observed in neighbouring molecules, carbon C(8)⋯C(11) (3.365(3)Å, 1 + x, y, z) and C(15)⋯C(18) (3.367(3)Å, 1 + x, y, z) were involved in the same. When viewed down a axis polymeric chains of M-1B molecules extend via C–H⋯O and Cl⋯Cl (3.358(3)Å, 1/2 − x, 1/2 − y, −z) interactions (Fig. 4). A fascinating architecture with herring borne like structure for M-1B molecules can be noticed down c-axis (Fig. 5) consequent to slipped π–π stacking interactions (Fig. S24a†). A zig-zag chain of Cl⋯Cl contacts were further observed; the ∠Cl⋯Cl⋯Cl being nearly 90° as shown in Fig. S24b.†
Table 2 Hydrogen bond geometries for M-1B, M-2B and M-4B
| Com. |
Sr. no. |
D–H⋯A |
D–H (Å) |
H⋯A (Å) |
D⋯A (Å) |
∠D–H⋯A (°) |
| −2 + x, 1 + y, z. 3 − x, 1/2 + y, 1/2 − z. 1 − x, 1 − y, 1 − z. x, 1/2 − y, −1/2 + z. x, 2.5 − y, 1/2 + z. x, 1 + y, z. |
| M-1B |
1 |
C(5)–H(5)⋯Cl(20)a |
0.950(2) |
2.797 (1) |
3.743(3) |
177.0(1) |
| 2 |
C(15)–H(15)⋯O(1)b |
0.951(1) |
2.399(2) |
3.285(3) |
154.8(1) |
| M-2B |
3 |
C(20)–H(20C)⋯O(1)c |
0.951(2) |
2.686(2) |
3.481(3) |
138.5(1) |
| 4 |
C(4)–H(4)⋯O(1)d |
0.951(2) |
2.695(1) |
3.356(3) |
127.2(1) |
| M-4B |
5 |
C(13)–H(13)⋯O(1)e |
0.951(3) |
2.623(2) |
3.382(4) |
137.1(2) |
| 6 |
C(21)–H(21C)⋯C(19)f |
0.980(3) |
2.830(3) |
3.685(4) |
146.1(2) |
 |
| | Fig. 4 Polymeric chains of M-1B via C–H⋯O and Cl⋯Cl interactions. | |
 |
| | Fig. 5 Molecular packing in M-1B down the c-axis. | |
M-2B molecule was in vicinity to four neighbouring molecules via C–H⋯O and π–π stacking interactions (Fig. S25†). M-2B molecules with head to tail orientation lead to a dimer via C–H⋯O interaction of C(19)–CH3 and carbonyl oxygen (C(2)–O(1)). The dimers also reveal π–π stacking interactions of C(2)⋯C(12) (3.216(2)Å, 1 − x, 1 − y, 1 − z), C(4)⋯C(14) (3.392(3)Å, 1 − x, 1 − y, 1 − z), C(8)⋯C(14) (3.386(3)Å, −x, 1 − y, 1 − z) and C(8)⋯C(16) (3.331(2)Å, 1 − x, 1 − y, 1 − z) (Fig. 6). Moreover, π–π stacking interactions from C(8)⋯C(14) extending as ladder structure of dimers, can further be noticed when viewed down c-axis of dimers. Neighbouring ladders of dimers were joined via C–H⋯O interactions. The “roller coaster” chain of dimers can be viewed down the c-axis (Fig. 7).
 |
| | Fig. 6 π–π stacked ladders in M-2B. | |
 |
| | Fig. 7 “Roller coaster” dimer chain in M-2B down the c-axis. | |
M-4B possess four neighbours surrounding the central molecules connected via C–H⋯O interaction and C⋯S interaction (Fig. S26†). The slipped C(18)⋯S(17) (3.458(3)Å, x,1 + y, z) interactions are supported by C–H⋯π interactions (C(19)⋯H(21C) = 2.830 Å, C(19)⋯C(21) = 3.685(4)Å, ∠C(21)–H(21C)⋯C(19) = 146.1°) (Fig. S26†). M-4B possess butterfly like molecular packing when viewed down the c-axis (Fig. S27†), where the plane of molecules makes an angle of nearly 45° to one another. Slipped C⋯S stacked polymeric chains further can be viewed down a-axis wherein the C–H⋯O interactions are present (Fig. 8).
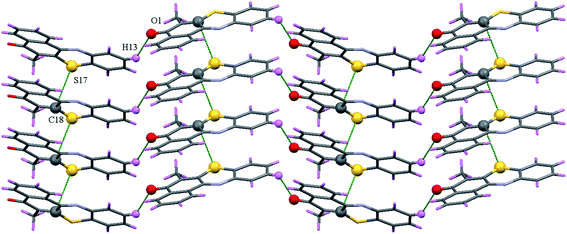 |
| | Fig. 8 C⋯S stacked polymeric chains of M-4B facilitated via C–H⋯O interactions. | |
Crystal structures showed the π–π stacking in addition to hydrogen bonding interactions in M-1B, M-2B and M-4B.
DFT investigations
Optimized structures of M-1B, M-2B, M-3B and M-4B from the M06-2x/6-31+G(d,p) density functional theory are shown in Fig. 9. Selected bond distances (in Å) are given along with the net atomic charges obtained from Hirschfield partitioning scheme in parenthesis. The structural parameters thus obtained agree fairly well with those from X-ray crystal data. To gain molecular insights for charge distribution governing dimer formation highest occupied molecular orbital HOMO were derived. An isosurface of −12.5 kcal mol−1 are portrayed in Fig. 10 in the M-1B to M-4B systems. It is evident that the electron-rich regions are largely localized on aromatic moieties. Remarkably enough, Fig. 10 points to diminutive charge localization around the phenolic ring, which partly has been attributed to −I effect from chlorine center. It may as well be inferred that π–π stacking contribute significantly along with hydrogen bonding interactions in the extended crystal network of M-1B, M-2B and M-4B.
 |
| | Fig. 9 M06-2x/6-31+G(d,p) optimized structures of M-1B, M-2B, M-3B and M-4B. Bond distances and net atomic charges (in parentheses) are given. | |
 |
| | Fig. 10 Frontier orbitals (isosurfaces of −12.5 kcal mol−1 in HOMO and LUMO). | |
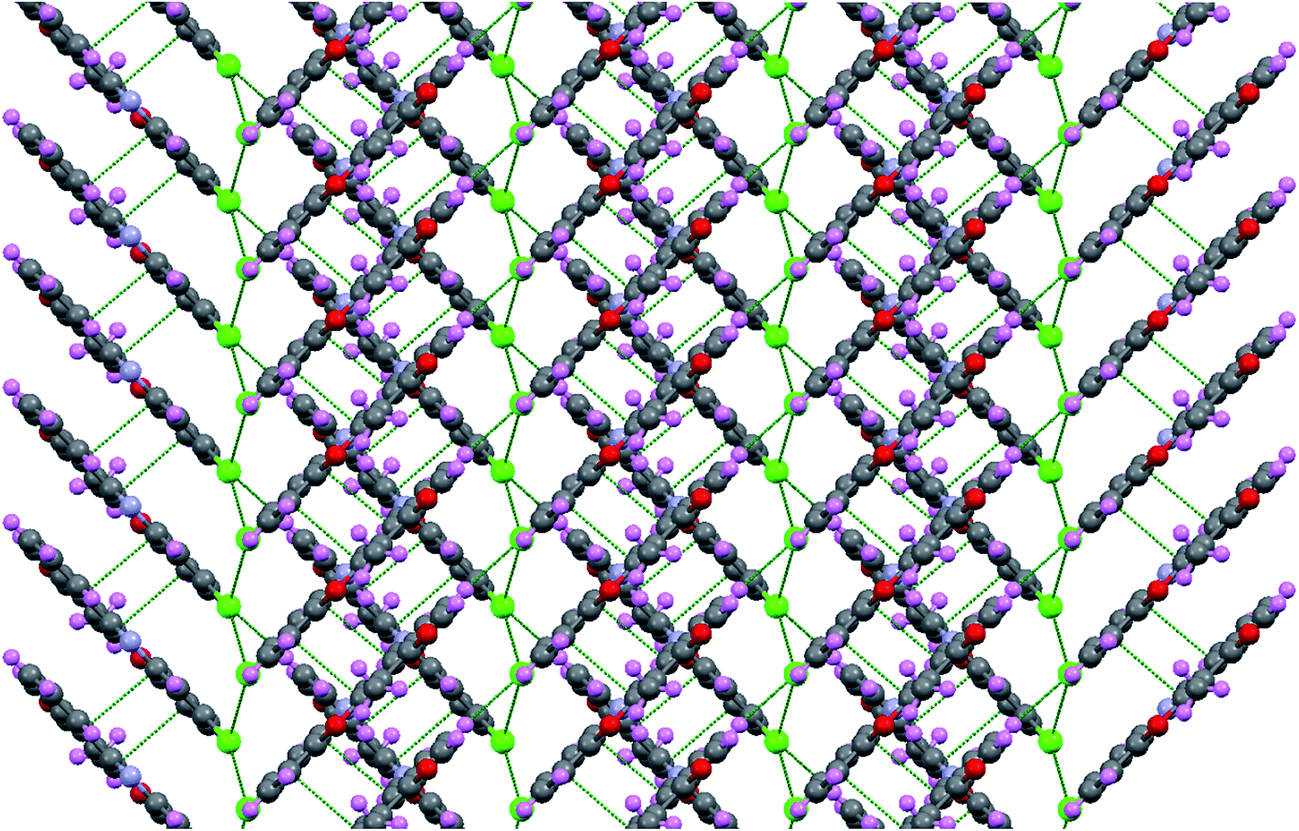
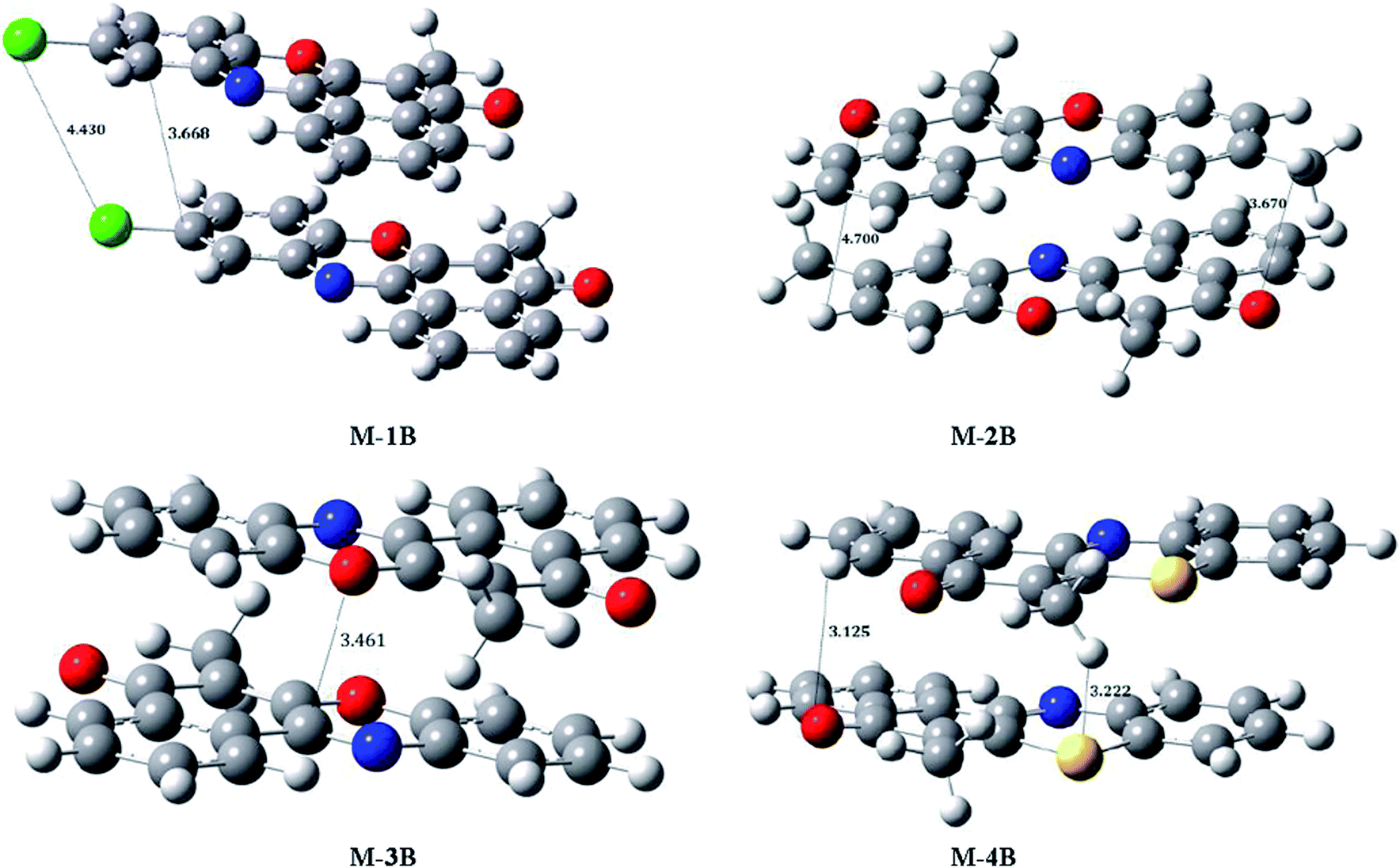
To understand interplay between stacking interactions and hydrogen bonding, we further delve into dimer formation accompanying M-1B to M-4B which has been shown in Fig. 11. The π–π stacking separations in the dimers range from 3.1–4.7 Å. Besides slipped π–π stacking interactions contributing to M-1B can be noticed from Fig. 11. The present theoretical calculations thus support inferences from the single crystal X-ray diffraction experiments. SCRF-PCM calculations further demonstrated that the solvent (DMSO) has no profound effect on the electronic structure.
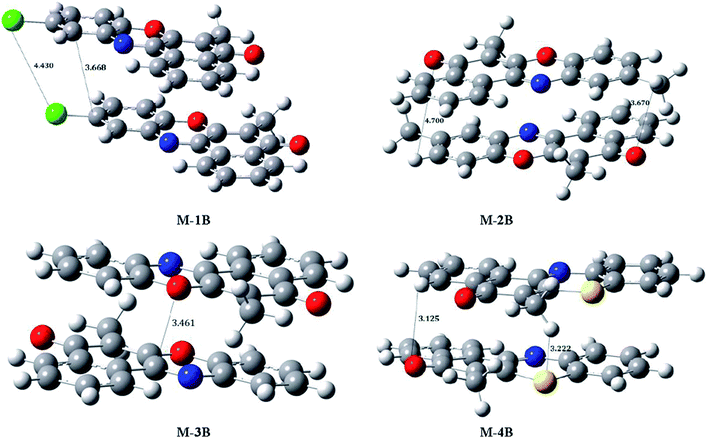 |
| | Fig. 11 M06-2x/6-31+G(d,p) optimized structures of M-1B, M-2B, M-3B and M-4B dimers. | |
Antiproliferative activity
The in vitro cytotoxicity of compounds M-1B to M-4B against MCF-7 (breast) and HeLa (cervical) cancer cell lines and normal skin cell cancer cell lines has been tested by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. All the compounds did not show any toxicity to normal skin cell; however, exhibit significant cytotoxicity in concentration dependent manner against MCF-7 and HeLa cells (Fig. S28–S30 in ESI†). The percent cell viability of MCF-7, HeLa and normal skin cell lines in the presence of complex M-1B to M-4B was measured in the concentration range of 5 μM to 25 μM, the compounds tested were found to be active at lower concentrations.
The IC50 values against MCF-7 and HeLa cell lines are presented in Table 3, which is comparable to cis-platin47 having IC50 of 16.7 ± 2.5 μM after 48 hours. Interestingly the M-2B is highly toxic against both MCF-7 and HeLa cell lines than the other compounds which possibly can be attributed to its stronger π–π stacking ability. Compounds M-1B to M-4B exhibits significant cytotoxicity against both cell lines viz. HeLa and MCF-7 by inducing apoptosis and can be envisaged as potential candidates for antitumor therapy. Cis-platin exhibits cytotoxic effect by covalent binding to DNA forming cis-platin-DNA adduct, which obstruct with DNA transcription, replication, and eventually leading cell death. It may thus be conjectured that the anticancer activities of M-1B to M-4B arise from partial intercalation or groove binding; the specific chemical structure and the nature of these compounds, on the other hand contribute significantly to enhanced cytotoxicity.
Table 3 Antiproliferative data (IC50) of M-1B to M-4B
| Compound |
MCF-7 (μM) |
HeLa (μM) |
| Each value reported here is a mean value of 3 independent experiments obtained after 48 hours. |
| M-1B |
21.52 ± 0.23a |
25.35 ± 0.13 |
| M-2B |
3.86 ± 0.33 |
4.35 ± 0.08 |
| M-3B |
8.32 ± 0.25 |
12.62 ± 0.12 |
| M-4B |
3.52 ± 0.21 |
13.75 ± 0.13 |
Cellular uptake
HeLa cells were treated with 25 μM of the respective compounds and after 24 h the cells were fixed and the images were captured using a fluorescence microscope. The cellular localization of compounds is monitored by fluorescence microscopy as these compounds emit green light on excitation with visible light. These compounds stain the nucleus of the cells (Fig. 12) and the mode of cell death was found to be apoptosis.
 |
| | Fig. 12 Fluorescence microscopy images of HeLa cells incubated (48 h) with 15 μM of compounds M-1B to M-4B. | |
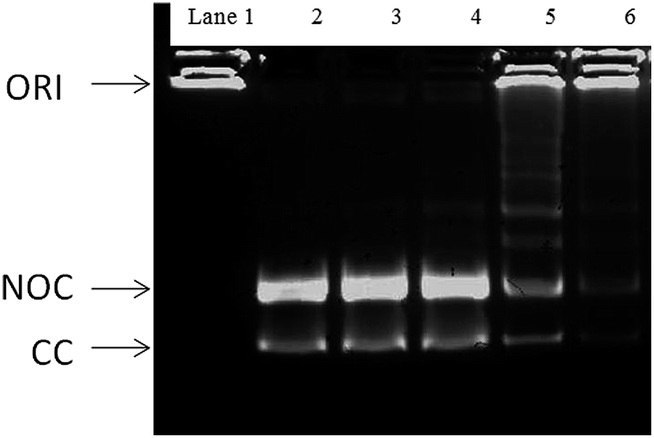
Fig. 13 depicts the gel decatenation assay analyzed for the compounds M-1B to M-4B. A wide network of many interlocked circular DNA forms the kDNA. Topoisomerase II, decatenates the kDNA which yields 2.5 kilobase relaxed decatenated kDNA monomers. Since smaller in size the monomers tend to run much faster on the gel.48 Topo-II act on the intact kDNA substrate to produce covalently closed circular decatenated kDNA (CC in Fig. 13). While its action on the nicked kDNA substrate formed during the isolation of kDNA produces nicked, open circular decatenated kDNA (NOC in Fig. 13). Noteworthy enough, compounds M-2B and M-4B are able to inhibit the Topo-II catalyzed decatenation of kDNA.
 |
| | Fig. 13 Lane 1: kDNA with no topoisomerase II; lane 2, decatenated kDNA control with no topoisomerase II in the assay buffer. Lane 3; Topo II + M-1B; lane 4; Topo II + M-3B; lane 5; Topo II + M-2B; lane 6; Topo II + M-4B. ORI, loading well origin; NOC, nicked, open circular decatenated kDNA; CC, covalently closed circular decatenated kDNA. The assay buffer contained 1.0 unit of topoisomerase II and 100 ng of kDNA. | |
Conclusions
Benzo[α]phenoxazines (M-1B to M-3B) and benzo[α]phenthiazine (M-4B) have been synthesized and characterized in this investigation. The precursor vitamin K3 was used in synthesis for the very first time with various derivatives of amino phenol and thioaminophenol. All synthesized planar polycycles are fluorescent. Molecular structures of M-1B, M-2B and M-4B revealed π–π stacking interactions as well as C–H⋯O, C–H⋯Cl and Cl⋯Cl non-covalent interactions. MO6-2X based density functional theory predicted electronic structures of M-1B to M-4B monomers as well as dimers agree well with the single crystal X-ray experiments wherein the π–π interactions are evident. The in vitro cytotoxicity of all compounds evaluated against MCF-7 (breast), HeLa (cervical) and normal skin cancer cell lines. All the compounds exhibits significant cytotoxicity against the MCF-7 and HeLa cell line by inducing apoptosis and further can be regarded as potential candidates for antitumor therapy. Moreover, and more important, compounds M-1B to M-4B showed very low cytotoxicity vs. normal skin cell lines. Cellular uptake studies on HeLa cell line of all compounds stain the nucleus of the cells and apoptosis causing the cell death. DNA catenation assay further suggested M-2B and M-4B are potential topoisomerase II inhibitors.
Experimental section
General materials and methods
The materials used viz. vitamin K3 (2-methyl-1,4-naphthoquinone), 2-aminophenol, 2-aminothiophenol were purchased from Sigma-Aldrich, 2-amino-4-methylphenol and 2-amino-4-chlorophenol were obtained from Across chemicals. The solvents used such as toluene, methanol are of analytical grade were purchased from Merck Chemicals. Solvents were distilled by standard methods49 and dried wherever necessary.
The FT-IR spectra of all the compounds were recorded between 4000–400 cm−1 as KBr pellets on SHIMADZU FT 8400 spectrometer (Fig. S1–S5 in ESI†). Mass of all compounds were determined by GC-MS 2010-eV (Make SHIMADZU) (Fig. S6–Fig. S9 in ESI†). Melting points of all compounds were determined using melting point apparatus (Make-METTLER) and were corrected using DSC (Differential Scanning Calorimetry) (Make-TA Q2000) (Fig. S10–S13 in ESI†). UV-Visible spectra of compounds were recorded on SHIMADZU UV 1650 in DMSO between 200 to 800 nm. The fluorescence spectra of the compounds were recorded on JASCO spectrofluorometer FP-8300. 1H,13CNMR and 2DgHSQCAD of compounds was recorded in CDCl3 on Varian mercury 500 MHz NMR instrument, TMS (tetramethylsilane) was used as the internal reference (Fig. S14–S21 in ESI†). Elemental analysis was performed on Elementar Vario EL III.
Synthesis
Synthesis of M-1B, M-2B, M-3B, M-4B. Vitamin K3, 1 g (5.8 mM) was dissolved in 25 mL of dry methanol, the solution was stirred for 20 min. The solids of 2-amino-4-chlorophenol (0.834 g, 5.8 mM) for M-1B, 2-amino-4-methyl-phenol (0.850 g, 5.8 mM) for M-2B, 2-aminophenol (0.872 g, 5.8 mM) for M-3B, 2-aminothiophenol (0.621 mL, 5.8 mM) for M-4B were dissolved in 15 mL of dry methanol independently; each solution was added drop wise to the solution of vitamin K3 with continuous magnetic stirring. The color of solution turns dark red to brown. The reaction mixtures of M-1B to M-3B were stirred at room temperature (26 °C) for 24 hours and are further refluxed for 60, 32 and 28 hours respectively for M-1B, M-2B and M-3B. The reactions were monitored by thin layer chromatography with 2% methanol in toluene (methanol:toluene (2:8)) as a mobile phase. Orange band was separated for all three compounds as a major product which was dried in air followed by vacuum. The X-ray quality crystals for M-1B and M-2B were obtained after recrystallization in toluene. In M-4B a dark orange coloured precipitate was obtained within 5 minutes of mixing the reactants at room temperature 26 °C, which showed single spot on TLC. This precipitate further filtered and dried in vacuum. An X-ray quality crystal of M-4B was obtained after recrystallization of the powder product in toluene.
Characterization
10-Chloro-6-methyl-7a,11a-dihydro-5H-benzo[a]phenoxazin-5-one; M-1B. Dark orange solid, yield: 0.492 g (29%), mp 222.58 °C. Anal. data calc. for C17H10ClNO2: C, 69.05; H, 3.41; N, 4.14; found: C, 69.19; H, 3.78; N, 4.56. FT-IR (KBr, νmax/cm−1): 3063, 3030, 2953, 2914, 2852, 1630, 1574, 1523, 1462, 1413, 1381, 1348, 1306, 1259, 1240, 1151, 1095, 1082, 962, 922, 871, 825, 783, 736, 682, 642, 588, 538, 497, 437. 1H NMR (500 MHz, CDCl3, δ/ppm): 2.221 (s, 3H), 7.264 (d, 1H, J = 8.00 Hz), 7.794 (d, 1H, J = 8.00 Hz), 7.239 (s, 1H), 7.756 (2, 2H, J = 8.00 Hz), 8.239 (d, 1H, 7.25 Hz), 8.646 (d, 1H, J = 8.00 Hz). 13C NMR (500 MHz, CDCl3, δ/ppm): C(1) = 124.85, C(2) = 132.11, C(3) = 132.21, C(4) = 126.32, C(4A) = 131.99, C(5) 183.71, C(6) = 116.99, C(6A) 143.41, C(7A) = 133.50, C(8) = 117.04, C(9) = 130.84, C(10) = 129.93, C(11) = 129.16, C(11A) = 147.43, C(12A) = 148.52, C(12B) = 130.80, C(13) = 8.37. UV-Vis; (λmax/nm, DMSO): 450, 355, 297. GC-MS (EI) m/z: 295 (M+ + H).
6,10-Dimethyl-7a,11a-dihydro-5H-benzo[a]phenoxazin-5-one; M-2B. Dark orange solid, yield: 0.603 g (40%), mp 193.24 °C. Anal. data calc. for C18H13NO2: C, 78.53; H, 4.76; N, 5.09; found: C, 78.53; H, 5.11, N, 5.44. FT-IR (KBr, νmax/cm−1): 3066, 3030, 2916, 2739, 1630, 1585, 1532, 1458, 1373, 1338, 1309, 1280, 1267, 1134, 1093, 1039, 966, 871, 783, 700, 642, 588, 536, 464, 434. 1H NMR (500 MHz, CDCl3, δ/ppm); 2.230 (s, 3H), 7.205 (d, 1H, J = 7.50 Hz), 7.261 (d, 1H, J = 8.25 Hz), 7.589 (s, 1H), 7.729 (m, 2H, J = 8.25 Hz), 8.311 (d, 1H, J = 8.00 Hz), 8.769 (d, 1H, J = 8.00 Hz). 13C NMR (500 MHz, CDCl3, δ/ppm): C(1) = 124.70, C(2) = 131.74, C(3) = 131.85, C(4) = 126.30, C(4A) = 132.59, C(5)183.81, C(6) = 116.14, C(6A) = 142.86, C(7A) = 135.00, C(8) = 115.60, C(9) = 131.19, C(10) = 132.59, C(11) = 129.83, C(11A) = 141.36, C(12A) = 148.08, C(12B) = 132.07, C(13) = 8.36, C(13′) = 21.06. UV-Vis; (λmax/nm, DMSO): 445, 355, 293. GC-MS (EI) m/z: 279 (M+ + H).
6-Methyl-7a,11a-dihydro-5H-benzo[a]phenoxazin-5-one; M-3B. Dark orange solid, yield: 0.58 g (39%), mp 179.45 °C. Anal. data calc. for C17H11NO2: C, 78.15; H, 4.24; N, 5.36; found: C, 77.84; H, 4.24, N, 4.93. FT-IR (KBr, νmax/cm−1): 3061, 2995, 2914, 2850, 1936, 1836, 1628, 1591, 1523, 1452, 1379, 1352, 1313, 1230, 1184, 1149, 1089, 1028, 962, 893, 856, 763, 684, 642, 586, 538, 474, 436. 1H NMR (500 MHz, CDCl3, δ/ppm): 2.239 (3H, s), 7.270 (d, 1H, J = 8.00 Hz), 7.795 (d, 1H, J = 7.50 Hz), 7.324 (t, 1H, J = 7.25 Hz), 7.457 (t, 1H, J = 7.75 Hz), 7.773 (m, 2H, J = 8.00 Hz), 8.307 (d, 1H, J = 7.00 Hz), 8.935, (d, 1H, J = 7.50 Hz). 13C NMR (500 MHz, CDCl3, δ/ppm): C(1) = 124.71, C(2) = 131.79, C(3) = 131.90, C(4) = 126.21, C(4A) = 132.03, C(5) = 183.82, C(6) = 116.39, C(6A) = 144.85, C(7A) = 132.84, C(8) = 115.98, C(9) = 125.11, C(10) = 131.21, C(11) = 129.84, C(11A) = 147.47, C(12A) = 147.86, C(12B) = 131.10, C(13) = 8.24. UV-Vis; (λmax/nm, DMSO): 443, 356, 296. GC-MS (EI) m/z: 261 (M+ + H).
6-Methyl-5H-benzo[a]phenothiazin-5-one; M-4B. Dark orange crystal, yield: 1.30 g (80%), mp 178.06 °C. Anal. data calc. for C17H11NOS: C, 73.62; H, 4.00; N, 5.05; S, 11.16; found: C, 73.24; H, 4.22, N, 5.44. FT-IR (KBr, νmax/cm−1): 3055, 2982, 2899, 1628, 1593, 1541, 1439, 1310, 1244, 1184, 1095, 1031, 966, 891, 756, 688, 617, 569, 518, 449. 1H NMR (500 MHz, CDCl3, δ/ppm): 2.210 (s, 3H), 7.271 (d, 1H, J = 7.75 Hz), 7.405 (t, 1H, J = 7.50 Hz), 7.465 (t, 1H, 7.75 Hz), 7.914 (d, 1H, J = 8.00 Hz), 7.727 (m, 2H, J = 7.50 Hz), 8.316 (d, 1H, 7.50 Hz), 8.853 (d, 1H, J = 7.50 Hz). 13C NMR (500 MHz, CDCl3, δ/ppm): C(1) = 125.23, C(2) = 131.37, C(3) = 131.61, C(4) = 126.27, C(4A) = 133.88, C(5) = 179.62, C(6) = 124.00, C(6A) = 134.63, C(7A) = 133.88, C(8) = 125.73, C(9) = 127.78, C(10) = 129.70, C(11) = 133.21, C(11A) = 138.54, C(12A) = 144.99, C(12B) = 132.30, C(13) = 13.27. UV-Vis; (λmax/nm, DMSO): 258, 314, 380, 483. GC-MS (EI) m/z: 277 (M+ + H).
X-ray crystallography
An orange single crystal of M-1B, M-2B and M-4B were coated with perfluoropolyether, picked up with nylon loops and mounted in the nitrogen cold stream of the Bruker APEX-II diffractometer. Graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) from a Mo-target rotating-anode X-ray source was used. Final cell constants were obtained from least squares fits of several thousand strong reflections. Intensity data were corrected for absorption using intensities of redundant reflections with the program SADABS.50 The structure was solved readily by direct methods and subsequent difference Fourier techniques. The Siemens ShelXTL51 software package was used for solution and artwork of the structures, ShelXL97 (ref. 52) was used for the refinement. All non-hydrogen atoms were anisotropically refined and hydrogen atoms were placed at calculated positions and refined as riding atoms with isotropic displacement parameters. The crystallographic data is presented in Table 1.
Computational details
Structures of M-1B, M-2B, M-3B and M-4B were optimized within the framework of dispersion corrected M06-2x based density functional theory employing the Gaussian-09 program.53 The internally stored 6-31G basis set with diffuse functions being added on all the heavy atoms (designated as 6-31+G(d,p) basis) have been used for these optimizations.54,55 Stationary point structures thus obtained were confirmed to be local minima on the potential energy surface through vibrational frequency calculations (all the normal vibration frequencies were turned out to be real). Frontier orbital were subsequently analyzed for these local minima structures at the M06-2x/6-31+G(d,p) level of theory. To derive molecular insights accompanying extension of crystal networks in M-1B–M-4B their dimeric structures were optimized subsequently.
Cytotoxicity studies
Maintenance of cancer cell line. MCF-7, HeLa and normal skin cell lines were obtained from National Centre for Cell Sciences Repository, Savitribai Phule Pune University, Pune, India. The cells were maintained in DMEM media with 10% FBS and 0.1% antibiotic solution at 37 °C at 5% CO2 in the steri-cycle CO2 incubator with HEPA Class 100 filters, Thermo Electron Corporation.
Preparation of sample for cell line testing. The compounds were dissolved in 1% DMSO to obtain a solution of 1 mM concentration each. These samples were then filter sterilized using a 0.22 μM filter using syringe filter.
Testing of compounds on cell line. The cells were trypsinized using TPVG solution. 1 mL of 1 × 105 cells per mL of medium and dilutions of concentration 5, 10, 15 and 20 μM was added in 96 well plates (Tarsons) and kept in the CO2 incubator for 24, 48, 72 and 96 hours. All experiments were carried out in laminar flow hoods, Laminar Flow Ultraclean Air Unit, Microfilt, India. The cells were visualized using an Inverted Microscope, Olympus.
MTT assay. Solution of 5 mg mL−1 MTT was dissolved in PBS and filter sterilized using syringe filter. After incubation for the stipulated time 20 μL of MTT solution was added to 200 μL of cell content solution. The plate was incubated for 2 hours in the CO2 incubator. After incubation the media was removed. 200 μL of DMSO was added to each well to dissolve the crystals. The plate was kept into the 37 °C incubator for 5 min. Reading was taken on Plate reader, Thermo Electron Corporation and absorbance was measured at 540 and 620 nm.
Cellular uptake. For preparation for fluorescent images (HeLa cell line preinoculated with compounds, in 24 well plate), 100 μL of cell content was pipetted out and centrifuged at 5000 rpm for 10 minutes. Cells were briefly washed with PBS twice. The pellet was resuspended in 50 μL of PBS. Ten microliters of the suspended cells was placed on a clean glass slide and a cover slip was imposed in such a way that no air bubbles were obtained. Images were taken in a Carl Zeiss Axio Scope A1 fluorescence microscope with filter set no. 9 and excitation at 450–490 nm.
DNA decatenation assay. Reagents for the assay were purchased from TopoGEN (catalogue number 1001-1, TopoGEN, Inc., Port Orange, FL). Topoisomerase II was extracted according to the manufacturer's instructions from nuclei of HeLa cells (NCCS repository, India) and contained both isoforms as assessed by Western blotting. Calculation of specific topoisomerase II decatenation activity was based on complete decatenation of a given amount of catenated input DNA (100 ng kDNA) by a defined amount of nuclear extract containing the alpha and beta isoforms or of a defined amount of purified enzyme in a particular amount of time. The topoisomerase II activity from nuclei decatenated 0.07 ng catenated kDNA min−1 ng−1 extract. Purified topoisomerase II alpha (see above) decatenated 0.13 ng catenated kDNA min−1 ng−1. Vehicle (DMSO) or drug substance at concentrations to yield the desired end concentrations were added and 20 μL of the samples were preincubated for 5 minutes at 37 °C and 400 rpm in an Eppendorf thermomixer. Reactions were started by adding ATP (to 450 μM final concentration) to each sample and incubation was continued for 20 minutes. Reactions were terminated by placing the samples on ice and adding 4 μL stop/gel loading buffer. 20 μL of each sample were separated by 1% agarose gel electrophoresis. Gels were analyzed under a UV transilluminator and decatenated kDNA products were quantified using Alpha Ease FC (Fluor Chem 8900) image analysis software version 3.2.3 (Alpha Innotech, San Leandro, CA).
Acknowledgements
SSG is grateful the Department of Biotechnology, India for the financial support. DC thanks the University Grants Commission, New Delhi, India for the Junior Research Fellowship. SSR acknowledges the financial support from the Savitribai Phule Pune University, Pune, India for the award of research fellowship through the potential excellence scheme. Authors acknowledge Institute of Bioinformatics & Biotechnology (IBB), Savitribai Phule Pune University for Animal Tissue Culture facility.
References
- J. Jose and K. Burgess, Tetrahedron, 2006, 62, 11021–11037 CrossRef CAS PubMed.
- B. R. Raju, A. Daniela, G. Firmino, A. L. S. Costa, P. J. G. Coutinho and M. S. T. Gonçalves, Tetrahedron, 2013, 69, 2451–3246 CrossRef CAS PubMed.
- L. Yuan, W. Lin, K. Zheng, L. He and W. Huang, Chem. Soc. Rev., 2013, 42, 622–661 RSC.
- A. D. G. Firmino and M. S. T. Gonçalves, Tetrahedron Lett., 2012, 53, 4946–4950 CrossRef CAS PubMed.
- J. Xu, S. Sun, Q. Li, Y. Yue, Y. Li and S. Shao, Analyst, 2015, 140, 574–581 RSC.
- K. Tanabe, Z. Zhang, T. Ito, H. Hatta and S.-I. Nishimoto, Org. Biomol. Chem., 2007, 5, 3745–3757 CAS.
- M. Liu, M. Hu, Q. Jiang, Z. Lu, Y. Huang, Y. Tan and Q. Jiang, RSC Adv., 2015, 5, 15778–15783 RSC.
- R. Sun, W. Liu, Y.-J. Xu, J.-M. Lu, J.-F. Ge and M. Ihara, Chem. Commun., 2013, 49, 10709–10711 RSC.
- S. S. Bag, S. Ghorai, S. Jana and C. Mukherjee, RSC Adv., 2013, 3, 5374–5377 RSC.
- L. Kathawate, P. V. Joshi, T. K. Dash, S. Pal, M. Nikalje, T. Weyhermüller, V. G. Puranik, V. B. Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 397–405 CrossRef CAS PubMed.
- K. Hara, M. Okamoto, T. Aki, H. Yagita, H. Tanaka, Y. Mizukami, H. Nakamura, A. Tomoda, N. Hamasaki and D. Kang, Mol. Cancer Ther., 2005, 4, 1121–1127 CrossRef CAS PubMed.
- B. Blank and L. L. Baxter, J. Med. Chem., 1968, 11, 807–811 CrossRef CAS.
- F. Paula Carneiro, M. D. Carmo, F. R. Pinto, S. T. Coelho, B. C. Cavalcanti, C. Pessoa, C. A. D. Simone, I. K. C. Nunes, N. M. D. Oliveira, R. T. D. Almeida, A. V. Pinto, K. C. D. G. Moura, K. D. Moura, P. A. D. Silva and E. N. D. Silva Jr, Eur. J. Med. Chem., 2011, 46, 4521–4529 CrossRef PubMed.
- O. Wesolowaska, J. Molnar, G. Westman, K. Samuelsson, M. Kawase, I. Ocsovszki, N. Motohashi and K. Michalak, In Vivo, 2006, 20, 109–114 Search PubMed.
- T. Klabunde, H. M. Petrassi, V. B. Oza, P. Raman, J. W. Kelly and J. C. Sachhettini, Nat. Struct. Biol., 2000, 7, 312–321 CrossRef CAS PubMed.
- B. Moosmann, T. Skutella, K. Beyer and C. Behl, Biol. Chem., 2001, 382, 1601–1612 CrossRef CAS PubMed.
- C. Y. Soto, N. Andreu, I. Gilbert and M. Luquin, J. Clin. Microbiol., 2002, 40, 3021–3024 CrossRef CAS.
- B. Morak-Młodawska, M. Jeleń and K. Pluta, Pol. Merkuriusz Lek., 2009, 26, 671–675 Search PubMed.
- J. Molnar, R. Pusztal, A. Hever, S. Nagy and N. Motohashi, Anticancer Res., 1995, 15, 2013–2016 CAS.
- N. Motohashi, H. Sakagami, K. Kamata and Y. Yamamoto, Anticancer Res., 1991, 11, 1933–1937 CAS.
- A. Bolognese, G. Correale, M. Manfra, A. Lavechhia, O. Mazzoni, E. Novellino, V. Barone, A. Pani, E. Tramontano, P. L. Colla, C. Murgioni, I. Serra, G. Setzu and R. Loddo, J. Med. Chem., 2001, 45, 5205–5216 CrossRef PubMed.
- A. Bolognese, G. Correale, M. Manfra, A. Lavecchia, E. Novellino and S. Pepe, J. Med. Chem., 2006, 49, 5110–5118 CrossRef CAS PubMed.
- A. Alberti, A. Bolognese, M. Guerra, A. Lavecchia, D. Macciantelli, M. Marcaccio, E. Lovellino and F. Paolucci, Biochemistry, 2003, 42, 11924–11931 CrossRef CAS PubMed.
- A. Abe, M. Yamane and A. Tomoda, Anti-Cancer Drugs, 2001, 12, 377–382 CrossRef CAS PubMed.
- K. Shirato, K. Imaizumi, A. Abe and A. Tomoda, Biol. Pharm. Bull., 2007, 30(2), 331–336 CAS.
- B. S. B. Salomi, C. K. Mitra and L. Garton, Synth. Met., 2005, 155, 426–429 CrossRef CAS PubMed.
- J. C. Wang, Annu. Rev. Biochem., 1996, 65, 635–692 CrossRef CAS PubMed.
- J. C. Wang, Annu. Rev. Biochem., 1985, 54, 665–697 CrossRef CAS PubMed.
- J. M. Berger, S. J. Gamblin and J. C. Wang, Nature, 1996, 379, 225–232 CrossRef CAS PubMed.
- L. F. Liu, Annu. Rev. Biochem., 1989, 58, 351–375 CrossRef CAS PubMed.
- A. Y. Chen and L. F. Liu, Annu. Rev. Pharmacol. Toxicol., 1994, 34, 191–218 CrossRef CAS PubMed.
- S.-T. Zhuo, C.-Y. Li, M.-H. Hu, S.-B. Chen, P.-F. Yao, S.-L. Huang, T.-M. Ou, J.-H. Tan, L.-K. An, D. Li, L.-Q. Gu and Z.-S. Huang, Org. Biomol. Chem., 2013, 11, 3989–4005 CAS.
- J.-J. Liu, J. Sun, Y.-B. Fang, Y.-A. Yang, R.-H. Jiao and H.-L. Zhu, Org. Biomol. Chem., 2014, 12, 998–1008 CAS.
- M.-Y. Kim, W. Duan, M. Gleason-Gunman and L. H. Hurley, J. Med. Chem., 2003, 46, 571–583 CrossRef CAS PubMed.
- F. M. Deane, E. C. O'Sullivan, A. R. Maguire, J. Gilbert, J. A. Sakoff, A. McCluskey and F. O. McCarthy, Org. Biomol. Chem., 2013, 11, 1334–1344 CAS.
- J. G. Atwell, W. G. Rewcastle, C. B. Baguley and W. A. Denny, J. Med. Chem., 1987, 30, 664–669 CrossRef.
- M.-Y. Kim, W. Duan, M. Gleason-Guzman and L. H. Hurley, J. Med. Chem., 2003, 46, 571–583 CrossRef CAS PubMed.
- M. Duca, P. B. Arimondo, S. Léonce, A. Pierré, B. Pfeiffer, C. Monneret and D. Dauzonne, Org. Biomol. Chem., 2005, 3, 1074–1080 CAS.
- N. Vicker, L. Burgess, I. S. Chuckowree, R. Dodd, A. J. Folkes, D. J. Hardick, T. C. Hancox, W. Miller, J. Milton, S. Sohal, S. Wang, S. P. Wren, P. A. Charlton, W. Dangerfield, C. Liddle, P. Mistry, A. J. Stewart and W. A. Denny, J. Med. Chem., 2002, 45, 721–739 CrossRef CAS PubMed.
- Y. Ueno, Monatsh. Chem., 1982, 113, 641–643 CrossRef CAS.
- J. Koshitani and Y. Ueno, J. Prakt. Chem., 1983, 325, 165–167 CrossRef CAS PubMed.
- R. Patil, D. Chadar, D. Chaudhari, J. Peter, M. Nikalje, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 345–351 CrossRef CAS PubMed.
- S. Pal, M. Jadhav, T. Weyhermüller, Y. Patil, M. Nethaji, U. Kasabe, L. Kathawate, V. B. Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2013, 1049, 355–361 CrossRef CAS PubMed.
- O. Pawar, A. Patekar, A. Khan, L. Kathawate, S. Haram, G. Markad, V. Puranik and S. Salunke-Gawali, J. Mol. Struct., 2014, 105, 68–74 CrossRef PubMed.
- S. Salunke-Gawali, O. Pawar, M. Nikalje, R. Patil, T. Weyhermüller, V. G. Puranik and V. B. Konkimalla, J. Mol. Struct., 2014, 1056–1057, 97–103 CrossRef CAS PubMed.
- D. R. Thube, A. Todkary, S. Y. Rane, K. Joshi, S. P. Gejji, S. A. Salunke, F. Varret and J. Marrot, J. Mol. Struct.: THEOCHEM, 2003, 622, 211–219 CrossRef CAS.
- C. Tan, S. Wu, S. Lai, M. Wang, Y. Chen, L. Zhou, Y. Zhu, W. Lian, W. Peng, L. Ji and A. Xu, Dalton Trans., 2011, 40, 8611–8621 RSC.
- B. B. Hasinoff, A. M. Creighton, H. Kozlowska, P. Thampatty, P. William and J. C. Yalowich, Mol. Pharmaceutics, 1997, 52, 839–845 CrossRef CAS.
- D. D. Perrin and W. L. Armarego, Purification of Laboratory Chemicals, Pergamon Press, London, 1988, p. 260 Search PubMed.
- G. M. Sheldrick, SADABS, Bruker–Siemens Area Detector Absorption and Other Correction, Version 2008/1, University of Göttingen, Germany, 2006 Search PubMed.
- ShelXTL 6.14, Bruker AXS Inc., Madison, WI, USA 2003 Search PubMed.
- G. M. Sheldrick, SHELX-97 Program for crystal structure solution and refinement, University of Göttingen, Germany, 1997 Search PubMed.
- M. J. Frisch G. W. Trucks H. B. Schlegel G. E. Scuseria M. A. Robb J. R. Cheeseman J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Akajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian03, revision Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Chem. Theory Comput., 2005, 1, 415–432 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of M-1B to M-4B by FT-IR (Fig. S1–S5 and S22), GC-MS (Fig. S6–S9), DSC (Fig. S10–S13) and 1H,13C and 2DgHSQCAD NMR (Fig. S14–S21). Crystallography figures Fig. S23–S27, cytotoxicity assay Fig. S28–S30. Table S1 (FT-IR data) and crystallographic Table S2–S12. CCDC 1039204–1039206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra08496b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.